Pathologie des globules rouges
Nous diviserons ce chapitre sur la pathologie des globules rouges en 4 parties :
3 – Cytologie des anémies
Devant toute anémie le frottis sanguin doit être attentivement examiné pour rêchercher des anomalies des globules rouges mais aussi des autres éléments sanguins, tout particulièrement polynucléaires, monocytes et plaquettes. La cytologie sanguine peut être complétée par la cytologie médullaire mais celle-ci n'a d'intérêt que si le mécanisme de l'anémie est central. Un myélogramme n'a aucun intérêt dans une anémie périphérique.
Nous décrirons les principales anomalies cytologiques que l'on peut observer au cours des anémies d'abord dans les anémies périphériques puis dans les anémies centrales avec, pour ces dernières, référence aux anomalies médullaires qui leur sont associées.
1 – Cytologie des anémies périphériques
Les anémies périphériques sont de loin les plus fréquentes. Ce sont des anémies régénératives (la moelle est normale) dues à la disparition prématurée des hématies du sang, soit par hémorragie, soit par hémolyse (raccourcissement de la durée de vie des hématies). Les anomalies cytologiques constatées sont différentes selon ces deux mécanismes.
1 – Les anémies hémorragiques
Seules les anémies par petites hémorragies occultes, doublant, triplant ou plus les pertes de fer quotidiennes posent un problème étiologique et entrainent des anomalies cytologiques observables.
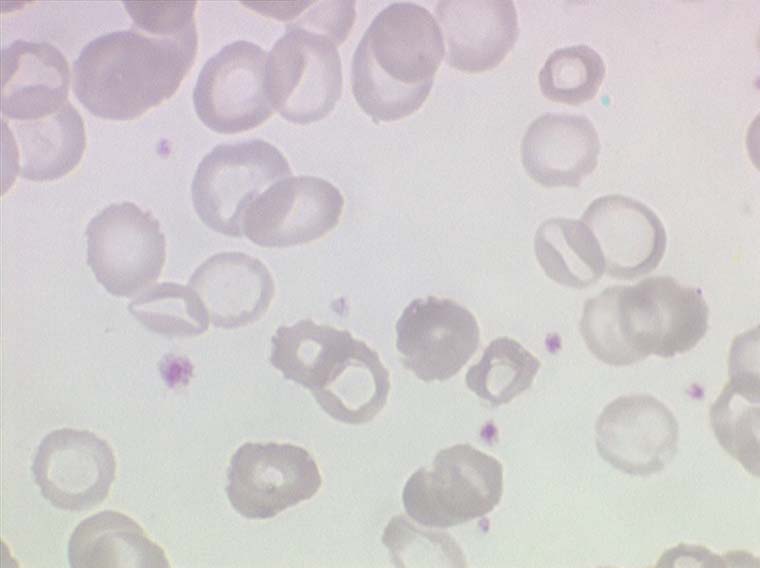
Dans ce type d'anémie le taux d'hémoglobine est fortement abaissé alors que le nombre de globules rouges est normal ou faiblement réduit (absence ou faible déglobulisation). La caractéristique principale de l'anémie hémorragique est d'être fortement microcytaire (VGM souvent inférieur à 80µ3) et hypochrome (TGM < 26µµg).
L'anémie hémorragique est certes par définition « régénérative » (élévation du taux de réticulocytes), mais le diagnostic étant souvent tardif cette régénération médullaire commence à s'épuiser et il est fréquent que le taux de réticulocytes soit proche de la normale.
Le fait principal à l'examen du frottis de sang est la microcytose et l'hypochromie des globules rouges, parfois à un degré tel que les hématies sont réduites à un mince anneau (annulocytes). On peut observer aussi quelques cellules cibles.
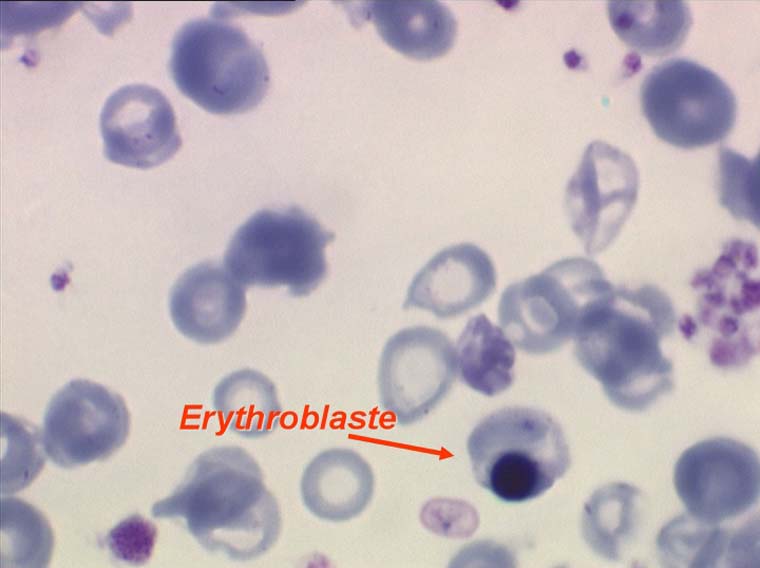
La réaction médullaire est responsable d'une relative thrombocytose et polynucléose. Le taux des plaquettes et des polynucléaires peut être au dessus des normes admises ou, à tout le moins, dans la partie haute de ces normes. Plus rarement cette réaction médullaire se traduit sur lame par une petite myélémie ou érythromyélémie.
Dans tous les cas une étude du métabolisme du fer viendra compléter l'hémogramme.


2 – Les anémies hémolytiques
Les anémies hémolytiques sont les plus régénératives qui soient. En effet jouent dans le même sens d'une forte augmentation des réticulocytes, d'une part une réaction médullaire intense qui mobilise la quasi totalité des capacités prolifératives de la moelle osseuse, d'autre part le raccourcissement de la durée de vie de l'hématie qui relativise le pourcentage des réticulocytes (une durée de vie de 10 jours au lieu de 100 donne, ipso facto, un taux de réticulocytes de 10% au lieu de 1%). Ces deux phénomènes cumulés font que des taux de réticulocytes de 20%, 30% ou plus ne sont pas rares au cours d'une anémie hémolytique, surtout en période de crise aiguë. Cette augmentation du taux de réticulocytes est en fait proportionnelle à la durée de vie de l'hématie, l'activité médullaire accrue ne pouvant expliquer que moins de 5% d'augmentation. Ceci implique qu'un taux de réticulocytes supérieur à 6% est toujours le reflet d'une hémolyse.

En dehors de cette forte régénération les autres caractéristiques de l'anémie hémolytique sont :
- une macrocytose relative qui est en fait proportionnelle au taux de réticulocytes qui ont, naturellement, un volume aux alentours de 120µ3,
- une augmentation des polynucléaires et des plaquettes qui est le reflet de la forte activité médullaire,
- une myélémie ou érythromyélémie fréquente de même cause.
L'examen du frottis sanguin est fondamental car des anomalies spécifiques des globules rouges peuvent, à peu de frais, orienter vers des étiologies précises.

Les causes d'anémies hémolytiques se divisent en deux types de maladies, congénitales et acquises. L'âge de survenue de la maladie joue donc un grand rôle dans ce diagnostic. Cependant si une anémie hémolytique chez le jeune enfant ou l'adolescent a toute chance d'être congénitale, une hémolyse, chez l'adulte, n'est pas toujours acquise car une anomalie génétique peut avoir une expression tardive.
Cytologie des anémies hémolytiques congénitales
Le globule rouge étant un petit sac (membrane) contenant des enzymes glycolytiques pour sa survie et de l'hémoglobine pour sa fonction (cf. physiologie du globule rouge), chacune de ces composantes peut être le siège d'une anomalie génétique transmissible.
1 – Les principales anomalies de la membrane rencontrées en France sont la maladie de Minkowski-Chauffard et l'elliptocytose familiale.
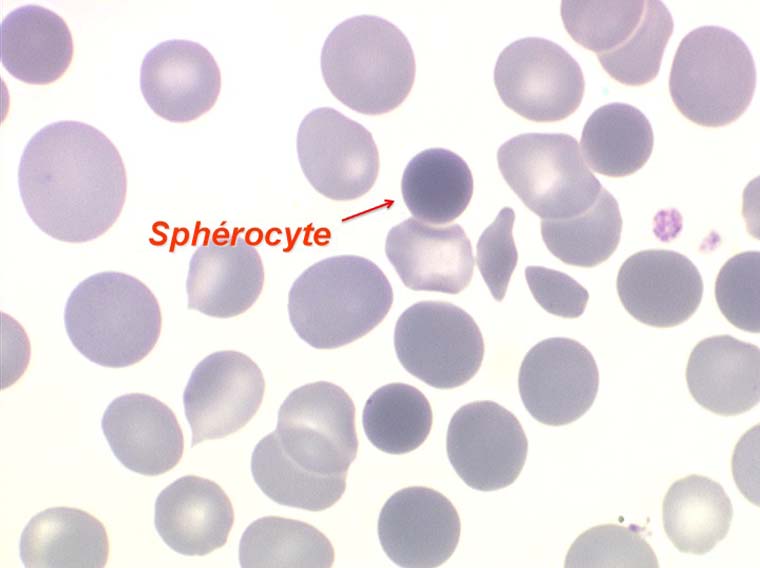
La maladie de Minkowski-Chauffard est caractérisée par la présence sur le frottis de sang de sphérocytes, globules rouges parfaitement sphériques apparaissant comme des hématies rondes (tracées au compas), denses, foncées (presque noires) de petit diamètre (alors que leur volume, donc leur VGM, est normal). Ces hématies sous pression, gonflées de liquide, sont fragiles et éclatent in vitro lorsqu'on les met dans une solution de moindre concentration osmotique (méthode diagnostique).
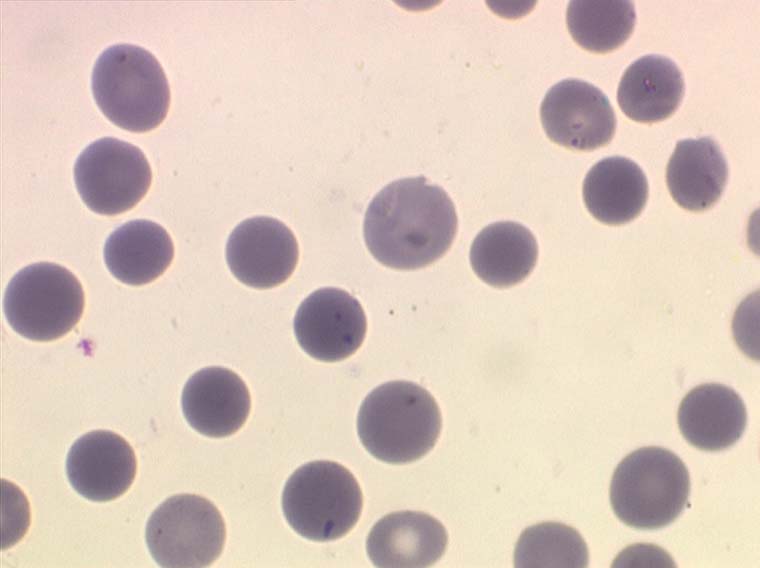
L'elliptocytose familiale est caractérisée par la présence sur le frottis de sang de globules rouges de forme ovalaire (ovalocytes). Le contour de ces cellules ovalaires est des plus réguliers, les bords ne présentent pas de crénelures, les extrémités ne sont pas acuminées. Il s'agit habituellement d'une maladie hétérozygote (un seul des parents a transmis le gène malade) et dans ce cas 50% des hématies sont ovalaires. Les très rares formes homozygotes ont 100% d'hématies ovalaires et elles seules peuvent faire des poussées d'hémolyse avec anémie.
2 – Les principaux déficits enzymatiques congénitaux (enzymopathies) rencontrés en France sont le déficit en G-6-PD (Glucose-6-phosphate déshydrogénase) et en PK (Pyruvate kinase). Ils n'entrainent pas d'anomalie morphologiques spécifiques des hématies, notamment pas de sphérocytose. Il est fréquent cependant de noter une certaine inégalité de formes des hématies (poïkylocytose).
3 – Les principales anomalies congénitales de l'hémoglobine rencontrées (hémoglobinopathies) sont la drépanocytose et les thalassémies
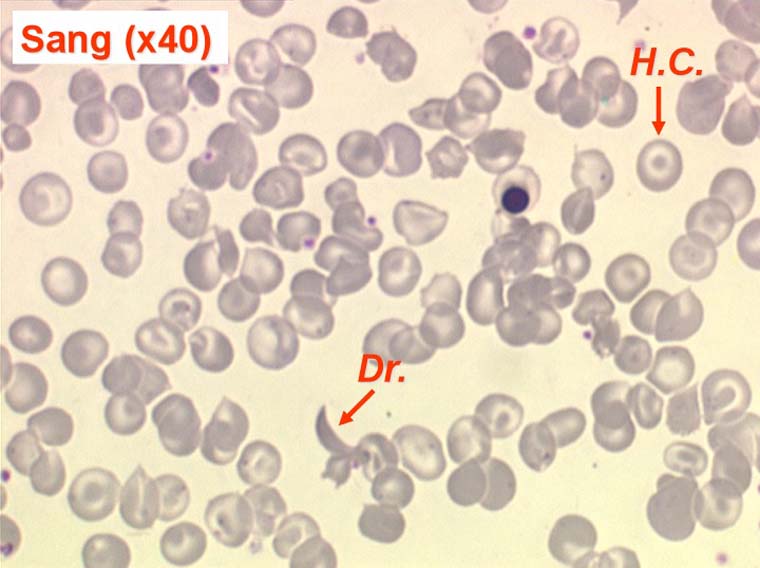
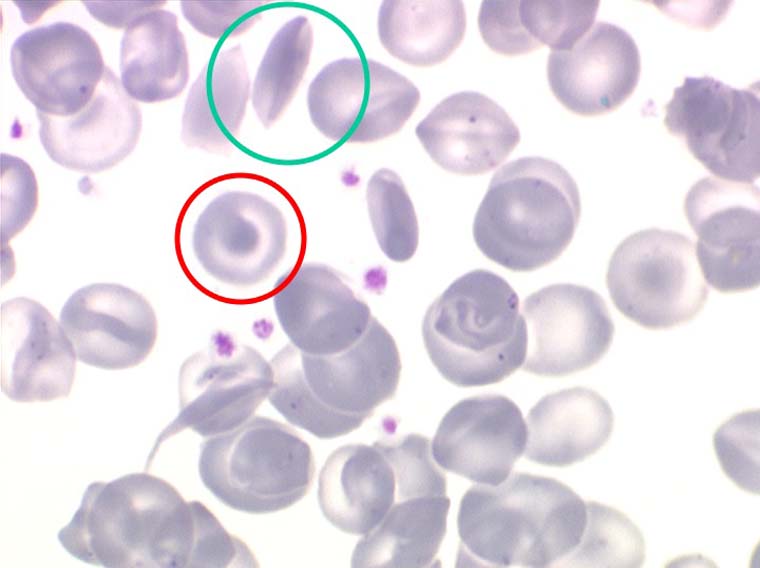
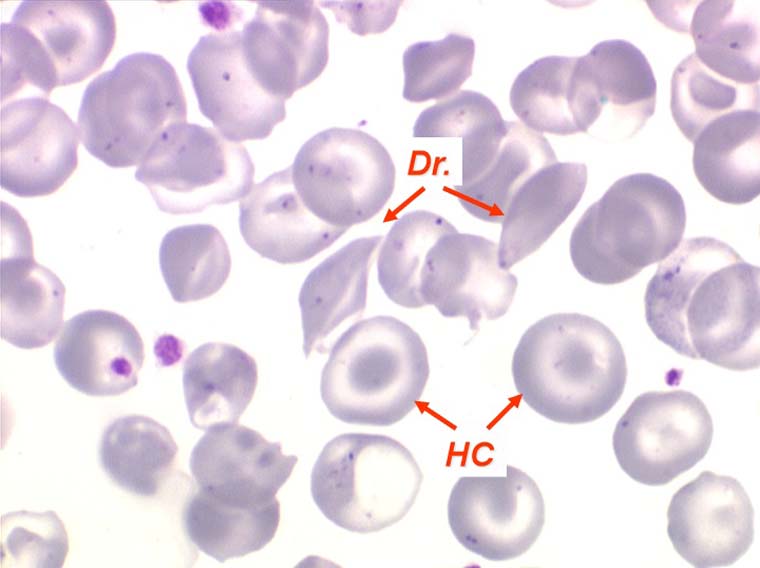
La drépanocytose (ou hémoglobinose-S) est la plus répandue des hémoglobinopathies, touchant des millions de noirs américains et africains et est une maladie grave et invalidante dans sa forme homozygote. Elle est due à une mutation génétique d'un acide aminé dans les chaines de globine. Sa signature cytologique est la présence d'hématies falciformes sur le frottis de sang, en grande quantité dans les formes homozygotes, en petit pourcentage (de 2% à 5%) dans les formes hétérozygotes.
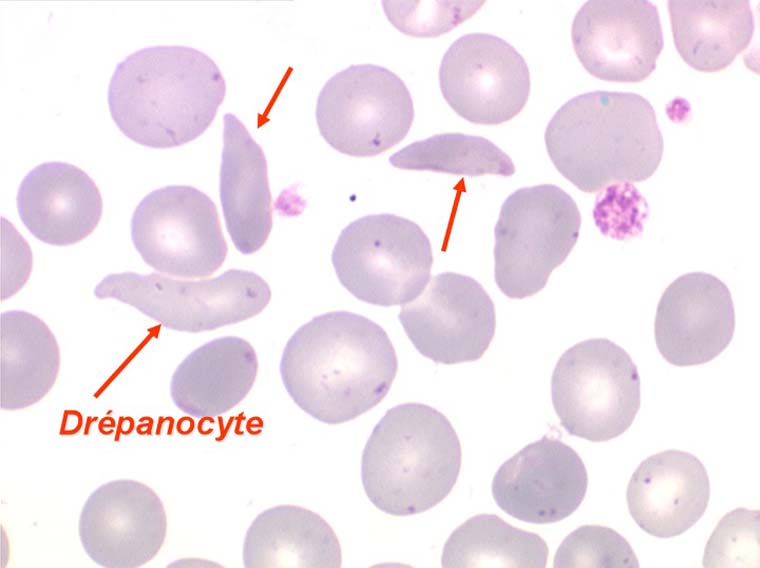
Ces hématies sont ovalaires mais à bord souvent crénelé et à extrémités acuminées, en tétine, elles peuvent être incurvées d'ou leur comparaison à une lame de faucille.
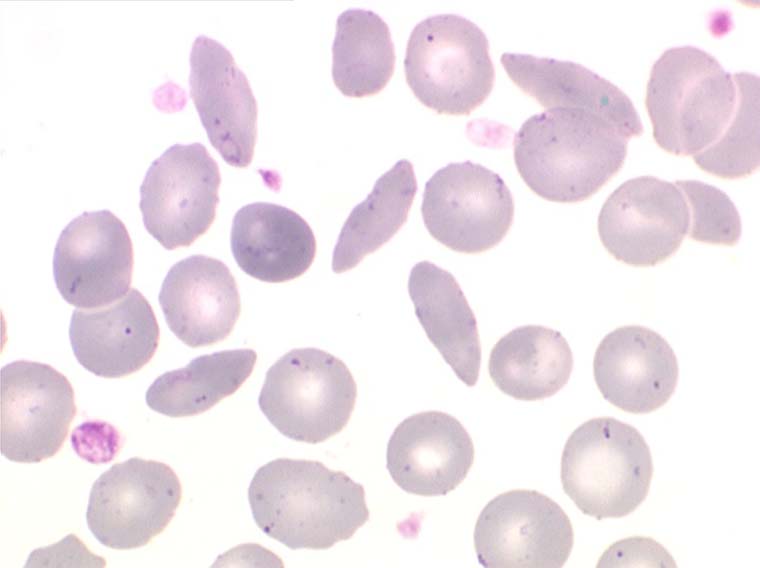
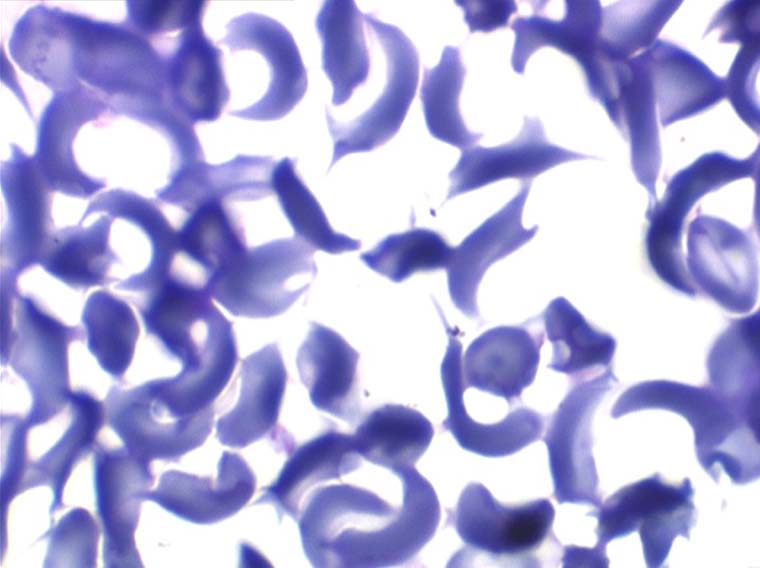
On peut créer artificiellement la déformation des hématies pathologiques in vitro en les privant d'oxygène. Sous l'effet de l'anoxie l'hémoglobine pathologique forme des pseudo cristaux qui déforment le globule rouge et lui donnent un aspect hérissé de spicules, en feuille de houx. C'est le phénomène de la falciformation. Ce phénomène existe aussi in vivo si se produit une anoxie tissulaire (altitude, fièvre, etc.) et ceci explique les microthromboses dans les vaisseaux capillaire et l'hémolyse qui s'en suit qui font toute la gravité de la maladie lors des crises drépanocytaires.
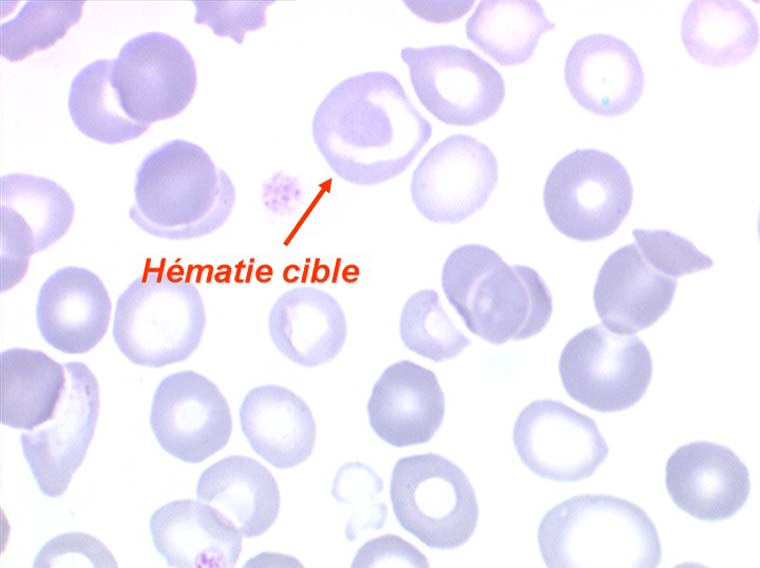
Les thalassémies sont dues à un défaut congénital de synthèse d'une des chaines de globine de l'hémoglobine, soit les chaines alpha (α) soit les chaines beta (β), les thalassémies portant sur les autres chaines (δ et γ) étant beaucoup plus rares. La répartition géographique des thalassémies est différente selon leur type, les β-thalassémies étant les plus fréquentes en Europe et autour du bassin méditerranéen. La signature cytologique des thalassémies est la présence d'hématies cibles sur le frottis sanguin, hématies en forme de cible de tir. Le pourcentage des hématies cibles est variable, plus important dans les formes homozygotes de la maladie.
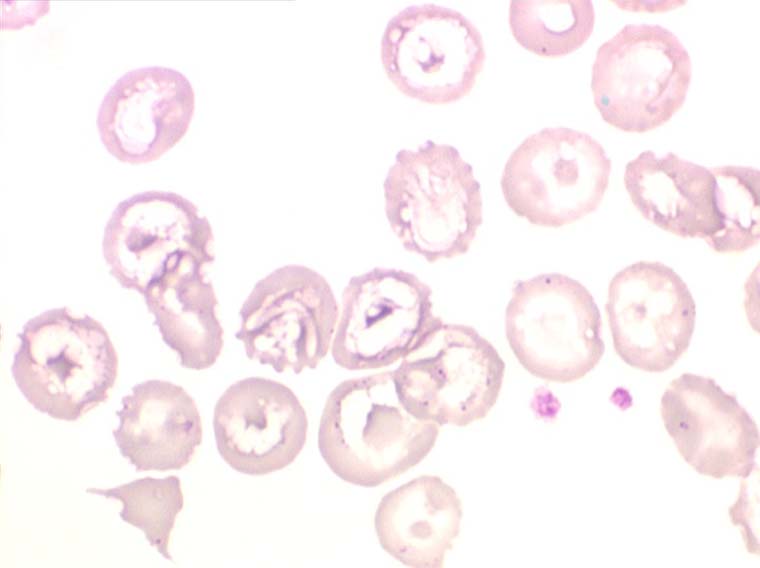
Seules les thalassémies homozygotes donnent des anémies sévères de mécanisme complexe où l'hémolyse n'est qu'une des composantes. Les thalassémies hétérozygotes sont beaucoup plus fréquentes et n'entraînent pas d'anémie, sinon très modérée, mais au contraire une polyglobulie hypochrome et microcytaire (voir le chapitre « polyglobulies secondaires »).
Compte tenu des brassages de populations, le double trait drépanocytaire et thalassémique peut coexister chez un même individu. Ceci s'observe dans les pays méditerranéens et dans les territoires français d'outre mer. La signature cytologique est alors la coexistence sur lame de drépanocytes et d'hématies cibles. Ces cas de double hétérozygotie, ou Drépano-Thalassémie, sont à dépister car ces sujets ont un risque de thrombose de l'artère centrale de la rétine qui peut être prévenu.
Cytologie des anémies hémolytiques acquises
Les anémies hémolytiques par auto-anticorps sont les plus fréquentes. Il s'agit d'une maladie auto-immune touchant le tissu immunitaire dont les cellules fonctionnelles (les lymphocytes) ne reconnaissent plus comme « soi » les propres constituants de l'organisme, notamment les globules rouges. Le diagnostic repose sur la mise en évidence des auto-anticorps par le test de Coombs, la lame de sang ne révèle pas grand chose si ce n'est une anisocytose et une polychromatophilie.
Lors des crises hémolytiques on peut observer des sphérocytes traduisant l'attaque de la membrane des hématies par les auto-anticorps.

La maladie des agglutinines froides est due à une élévation considérable du taux d'agglutinines froides qui existent naturellement à un taux très faible et sont actives à 4°. Ces agglutinines à taux élevé peuvent avoir une amplitude thermique accrue et être actives à des températures se rapprochant de la température corporelle. Il se produit alors des agrégats d'hématies in vitro qui forment des amas sur la lame. Ce phénomène rend la numération globulaire difficile voire impossible à la température du laboratoire. Il est responsable aussi chez le malade de microthromboses dans la microcirculation des extrémités (doigts, nez, lobes des oreilles) entrainant des nécroses.
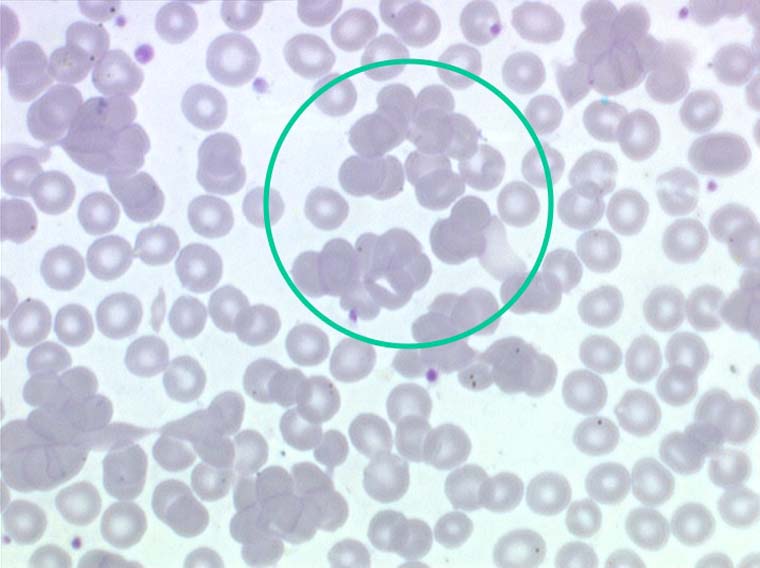
Les anémies hémolytiques avec schizocytose forment un groupe hétérogène d'hémolyse. Le schizocyte ou fragment d'hématie est un mode de mort naturel du globule rouge mais tous les globules rouges en fin de vie ne prenant pas l'aspect d'un schizocyte, le nombre de schizocytes sur une lame normale est faible et ne représente qu'une fraction de pourcentage. Au delà de 1% à 2% ce nombre devient pathologique. La morphologie des schizocytes est variable car sa formation à partir d'une hématie normale passe par plusieurs stades qu'illustrent les photographies suivantes. Ces stades chronologiques sont les suivants : apparition d'une vacuole dans l'hématie (stade 1), ouverture de la vacuole (stade 2), éversion des berges de la vacuole éclatée (stade 3 dit aussi aspect « en chapeau de gendarme »), aspect déchiqueté de l'hématie (stade 4 dit aussi « fragment d'hématie »). Chacun de ces aspects mérite le nom de schizocyte et coexiste sur une même lame.

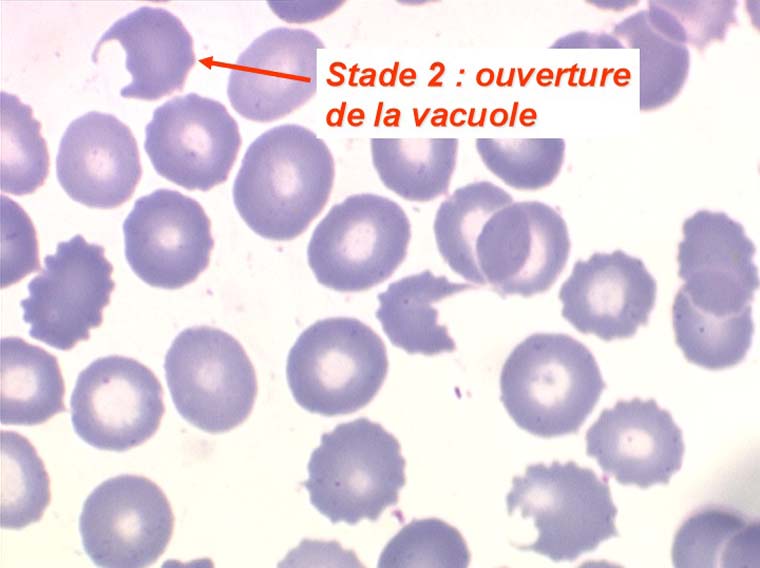
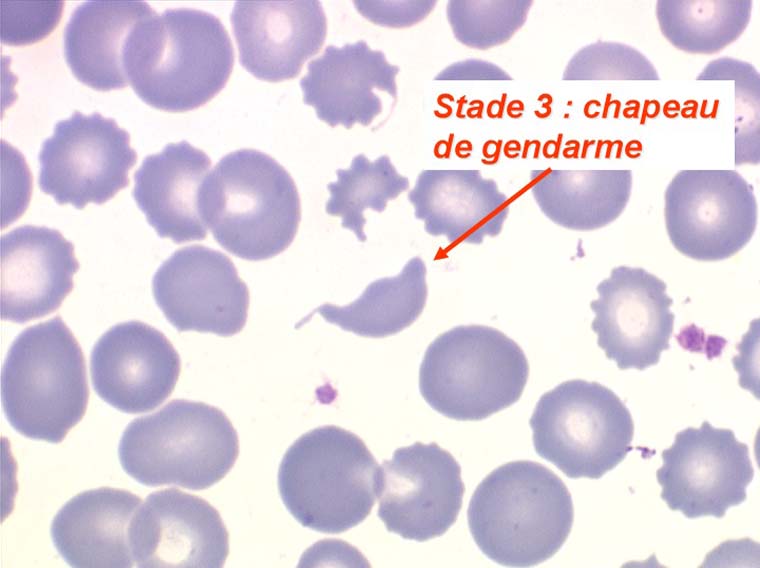
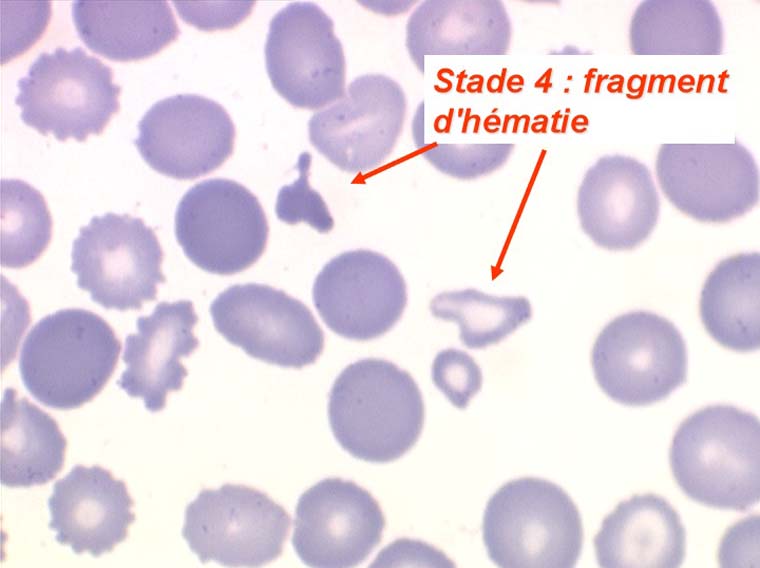
Une augmentation du pourcentage de schizocytes dans le sang s'observe dans plusieurs circonstances dont une est grave et constitue une urgence hématologique.
• Ce pourcentage augmente chez les malades ayant une prothèse vasculaire ou valvulaire cardiaque. Cette augmentation est liée à une fragilisation des globules rouges due à des phénomènes de turbulence au niveau de la prothèse. Le taux de schizocytes est inversement proportionnel à la qualité hémodynamique de la prothèse et permet de surveiller le fonctionnement de celle-ci.
• Beaucoup plus préoccupante est l'importante schizocytose (> 10%) que l'on observe dans le Purpura Thrombopénique et Thrombotique (PTT) ou maladie de Moschowitz. Cette schizocytose s'intègre dans le cadre d'une anémie hémolytique régénérative mais associée à une thrombopénie. L'examen de la lame est alors fondamental et doit déclencher une action thérapeutique immédiate compte tenu de la gravité du pronostic.
• On peut aussi observer une schizocytose sanguine associée à une anémie régénérative et à une érythromyélémie dans les métastases médullaires (cf. « Sangs pathologiques » → « Lignée granuleuse » → « Myélémies et syndromes myéloprolifératifs »).

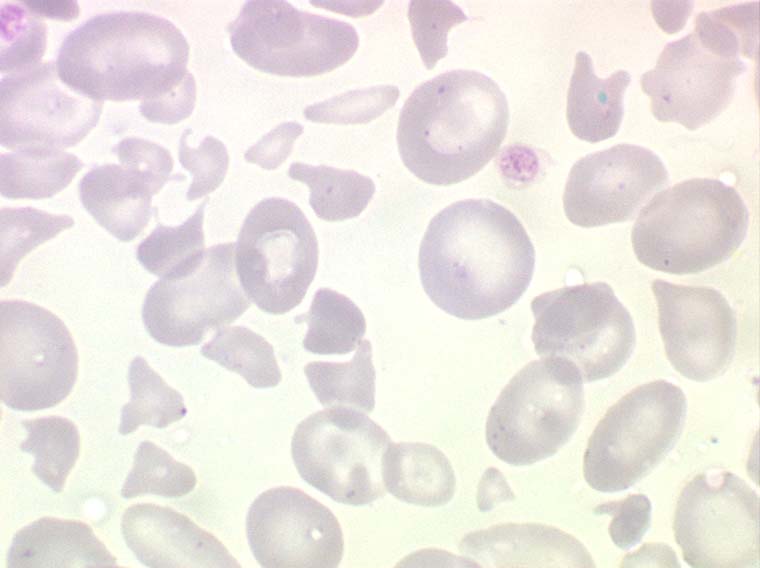
L'acanthocytose est caractérisée par des hématies crénelées en forme de feuille d'acanthe ou spiculées en forme d'oursin (on les appelle aussi échinocytes). Le plus souvent il s'agit d'un artefact (effet verre) sans aucune conséquence pathologique. Très rarement l'acanthocytose s'accompagne d'une hémolyse dans le cadre d'un alcoolisme sévère ou d'exceptionnelles dyslipoïdoses familiales.
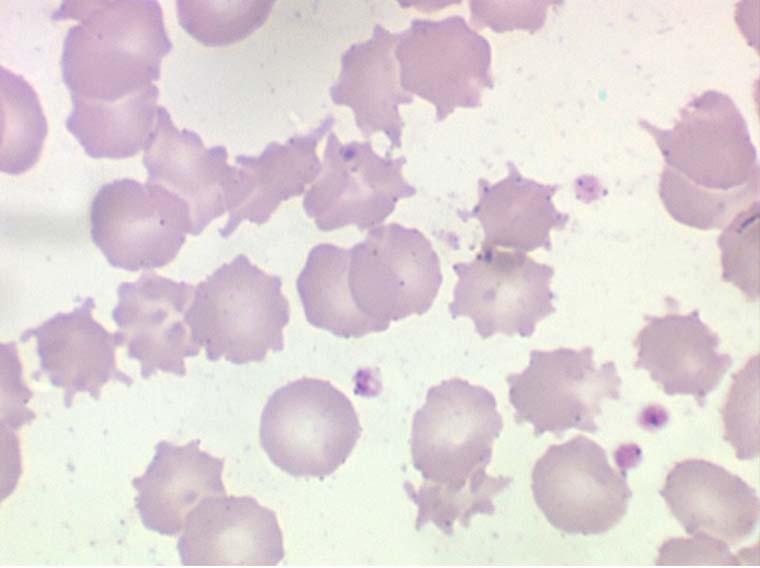
2 – Cytologie des anémies centrales
Les anémies centrales, c'est à dire dues à un dysfonctionnement de la moelle osseuse, peuvent être quantitatives (aplasie) ou qualitatives (myélodysplasie). Elles sont moins fréquentes que les anémies de mécanisme périphérique mais habituellement beaucoup plus graves.
Les anémies des aplasies médullaires
Sont dues à la forte diminution de la quantité de moelle active, quelque dizaines de grammes au lieu de plusieurs centaines de grammes. La moelle étant un organe extrêmement actif, en perpétuel renouvellement, elle est particulièrement sensible aux toxiques (notamment médicamenteux), aux radiations ionisantes et à certains virus. L'anémie des aplasies est quasi arégénérative (0% de réticulocytes), normocytaire et normochrome (les rares globules rouges fabriqués sont normaux) et surtout rarement isolée mais associée à une grande neutropénie (polynucléaires neutrophiles inférieurs à 1.000/µ3) et à une thrombopénie (plaquettes inférieures à 50.000/µ3) ce qui définit la pancytopénie.

L'examen de la lame de sang confirme la pancytopénie en montrant un sang quasi vide de polynucléaires et de plaquettes, les rares leucocytes rencontrés étant des lymphocytes qui, n'étant pas fabriqués par la moelle, sont les seuls présents dans le sang.
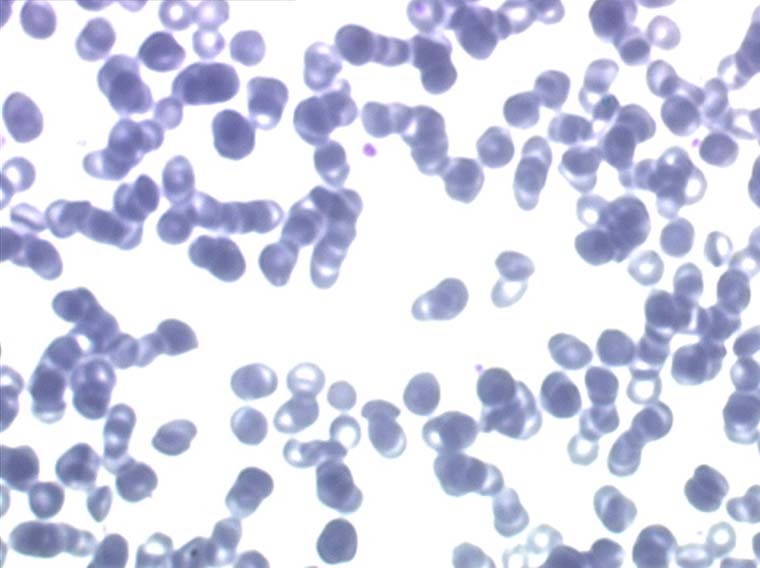
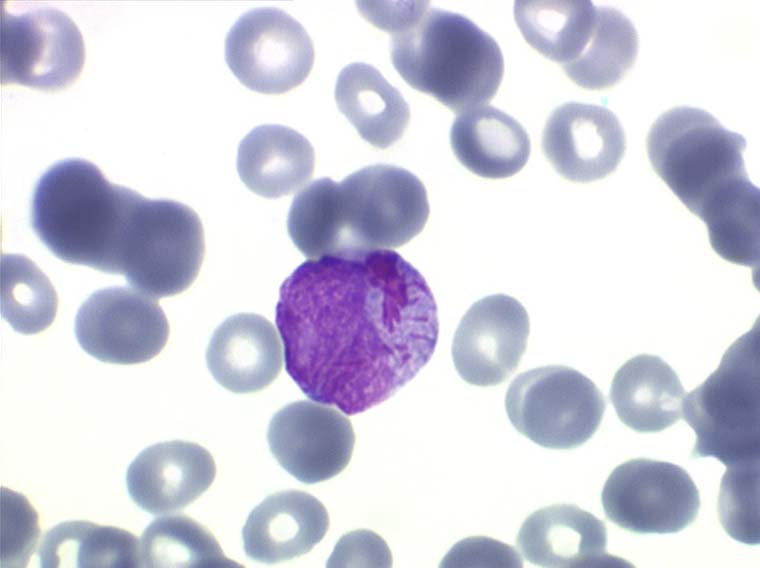
Il faut cependant être méfiant et examiner patiemment la lame de sang sur toute sa surface à la recherche de la moindre cellule anormale. En effet l'aplasie est l'un des deux termes obligés d'une leucémie aiguë, l'autre étant le passage dans le sang de blastes. Au début de certaines leucémies myéloïdes le nombre de cellules blastiques est faible et peut ne pas être détecté par le compteur de particules, seule l'aplasie est alors manifeste. C'est notamment le cas de la leucémie aiguë à promyélocytes (LAM 3) qui constitue une urgence hématologique. Devant toute aplasie il faut donc rechercher minutieusement d'éventuelles cellules blastiques. Dans le cas d'une leucémie à promyélocytes il s'agira de blastes hypergranuleux, contenant ou non des fagots de corps d'Auer.

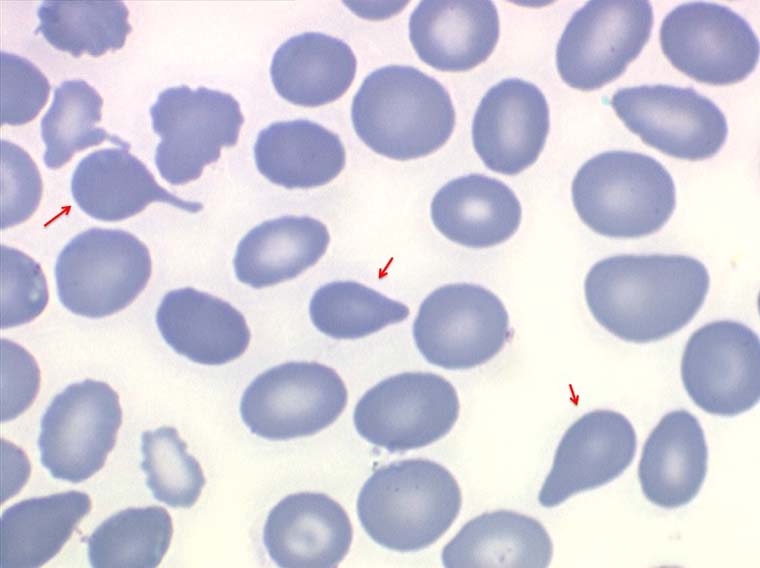
Une aplasie peut être due à une fibro-sclérose de la moelle. Dans ce cas la pancytopénie s'associe à une érythromyélémie qui peut être modérée mais surtout à des déformations des hématies, les dacryocytes, globules rouges en forme de larmes, de poires, de gouttes. Devant toute pancytopénie l'examen des globules rouges sur lame doit donc être un reflexe.
Bien évidemment devant une aplasie il est nécessaire de confirmer la vacuité de la moelle. L'examen de choix pour ce faire est l'étude anatomo-pathologique de la moelle prélevée par biopsie. Le myélogramme n'est pas utile car devant un aspect de moelle très pauvre on peut craindre une erreur technique lors du prélèvement. En revanche la ponction de moelle s'avère nécessaire si l'on suspecte une origine leucémique à l'aplasie. Le contraste est alors frappant entre un sang désertique et une moelle envahie de blastes.
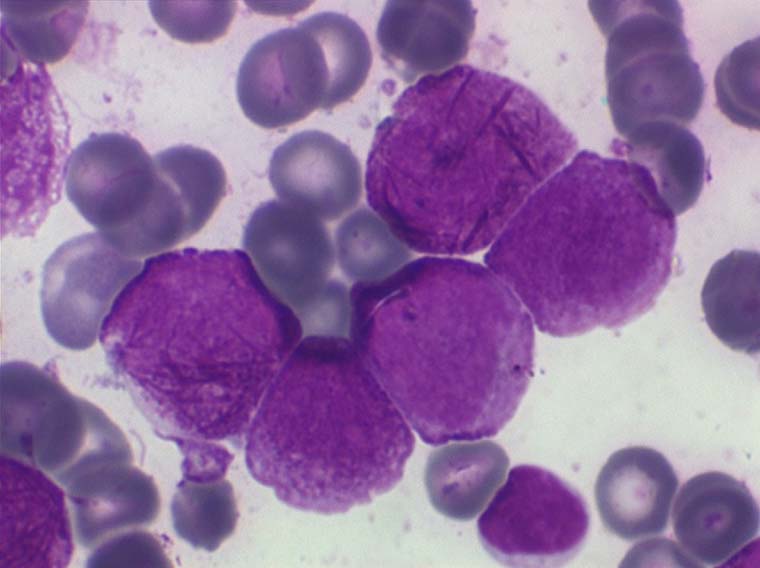
Les anémies des myélodysplasies
La myélodysplasie est une insuffisance qualitative de la moelle osseuse. Il s'agit d'une hématopoïèse inefficace (encore appelée dyshématopoïèse) qui se caractérise donc par une pancytopénie (anémie, neutropénie et thrombopénie) à moelle riche mais bloquée lâchant dans le sang périphérique des éléments de morphologie et de fonction anormales.
L'anémie des myélodysplasies est peu régénérative (taux de réticulocytes entre 0,1% et 0,5%) avec des constantes habituellement anormales, l'anomalie la plus fréquente étant une macrocytose et une anisocytose le plus souvent associée à une hypochromie. Devant toute anémie macrocytaire hypochrome peu régénérative il faut avoir le reflexe de penser à une myélodysplasie.


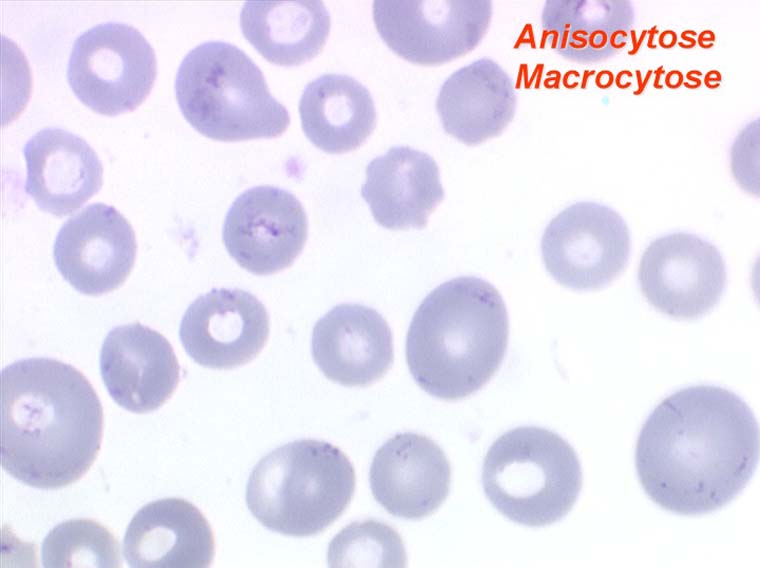
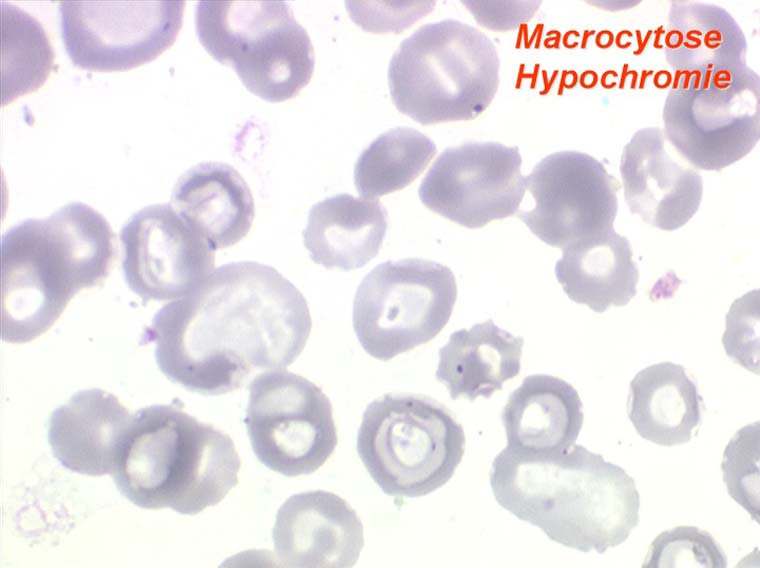
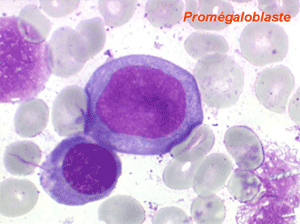
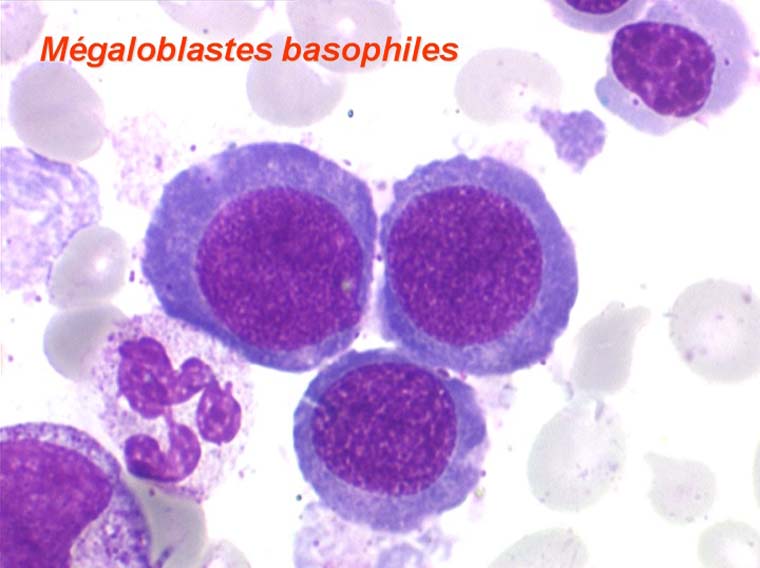
Dans certaines étiologies de myélodysplasie, les avitaminoses, la macrocytose peut être très importante. Les hématies sont énormes, de couleur uniforme, de forme volontiers ovalaire. On les appelle « mégalocytes ». Ceci s'observe surtout dans l'avitaminose B12 ou anémie de Biermer.

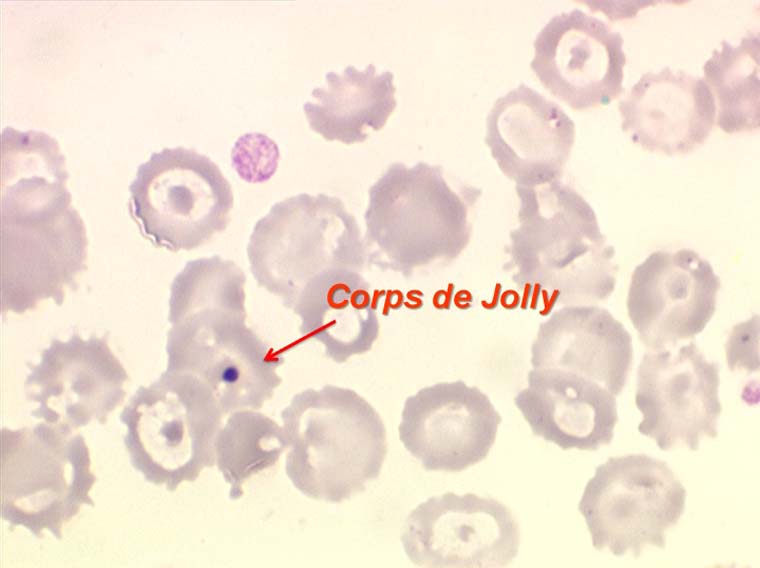
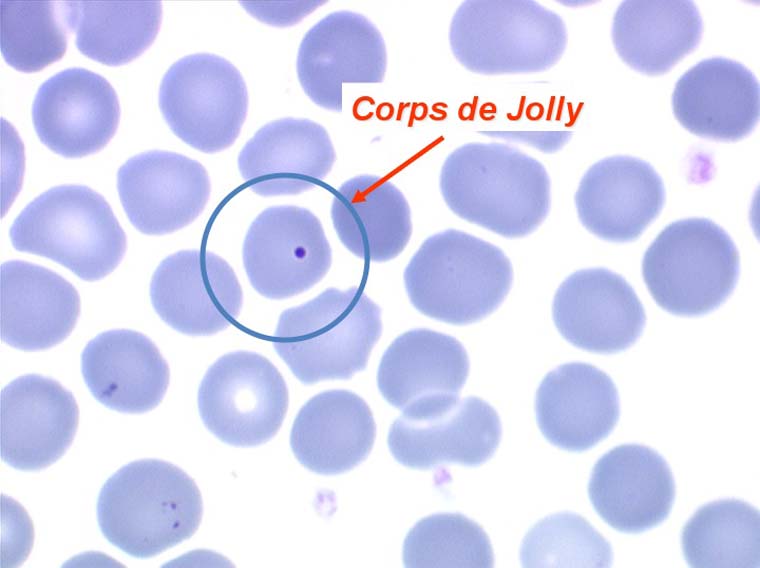
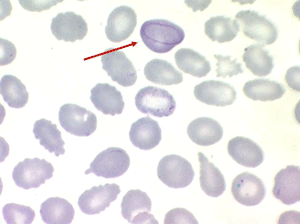
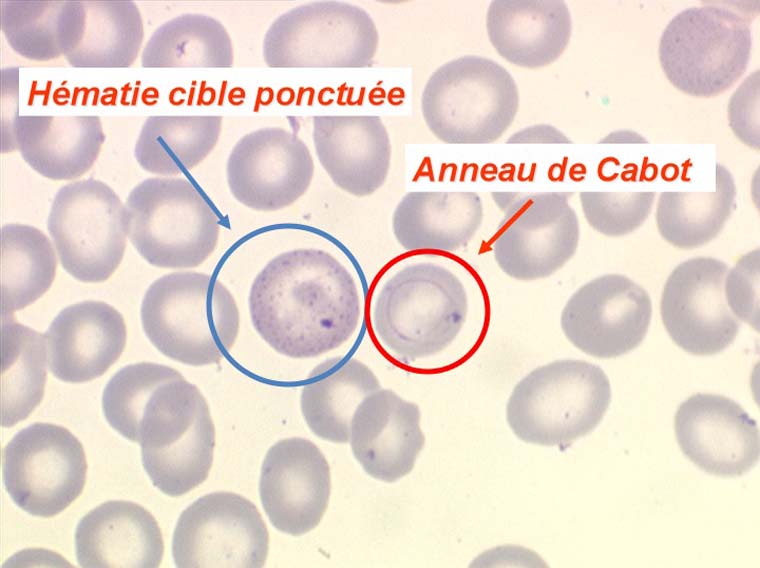
D'autres anomalies des globules rouges peuvent être observées au cours des myélodysplasies. Il s'agit d'inclusions intra-érythrocytaires telles que des corps de Jolly, des anneaux de Cabot (reste rigide de la membrane nucléaire de l'érythroblaste) et des ponctuations basophiles, anomalies coexistant parfois dans la même hématie. Ces inclusions font partie de « l'atmosphère myélodysplasique » qu'il faut savoir rechercher avec soin sur la lame de sang dès que l'on pense à ce diagnostic, notamment, répétons le, devant une anémie peu régénérative, macrocytaire et hypochrome.
Les anomalies des globules rouges ne sont pas les seules observées au cours des myélodysplasies, les polynucléaires et les plaquettes, fabriqués par cette moelle malade, ont aussi des anomalies telles que dégranulation, anomalies de lobulation, plaquettes géantes et micro mégacaryocytes circulants. De même il est fréquent d'observer une petite myélémie ou érythromyélémie, avec même présence de quelques myéloblastes ou promyélocytes. Ces anomalies associées seront décrites dans le chapitre de la pathologie de la lignée granuleuse.
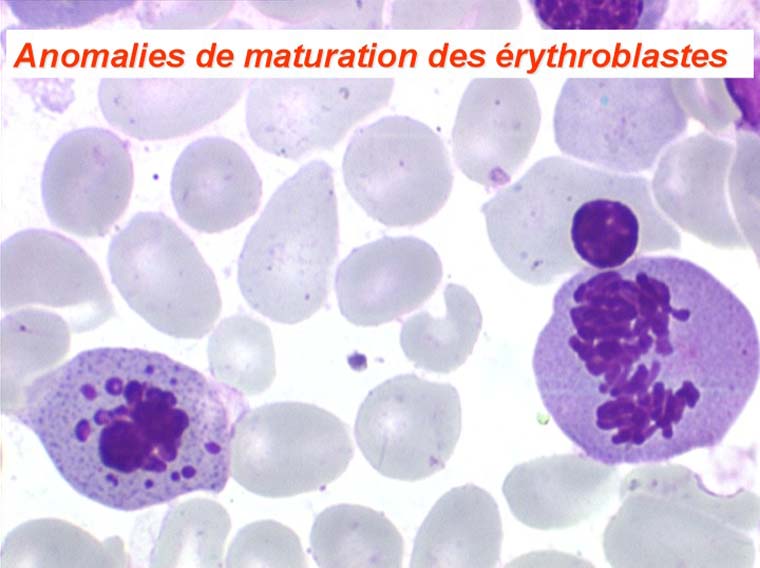
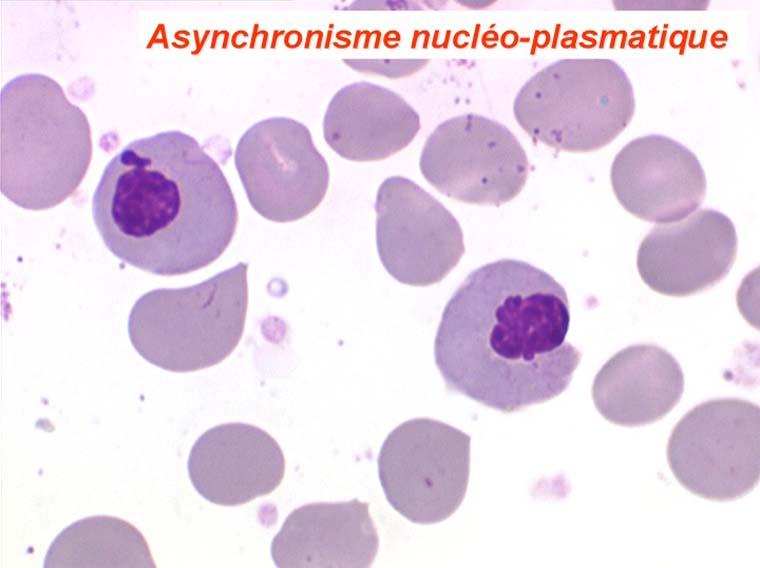
Les anomalies morphologiques de la moelle osseuse sont aussi très importantes. Nous ne ferons que citer ici celles portant sur la lignée érythroblastique. Les principales anomalies observées dans cette lignée sont la richesse accrue en érythroblastes, une tendance au gigantisme cellulaire (mégaloblastose), des anomalies nucléaires (chromatine perlée des stades les plus jeunes, fragmentation anormale des noyaux des stades les plus âgés), un asynchronisme de maturation entre le noyau (qui reste jeune) et un cytoplasme (qui vieillit plus vite). À ces anomalies des érythroblastes s'associent des anomalies des autres lignées médullaires qui seront décrites dans le chapitre de la pathologie de la lignée granuleuse.
Retour haut de page