
Lignée granuleuse
Ce chapitre sur la ligné granuleuse est divisé en 6 parties :
(le chapitre 4 - Leucémies aiguës myéloïdes a été fractionné en deux pour optimiser le temps de chargement des photos)
3 –Myélémies et syndromes myéloprolifératifs
1 – Les myélémies
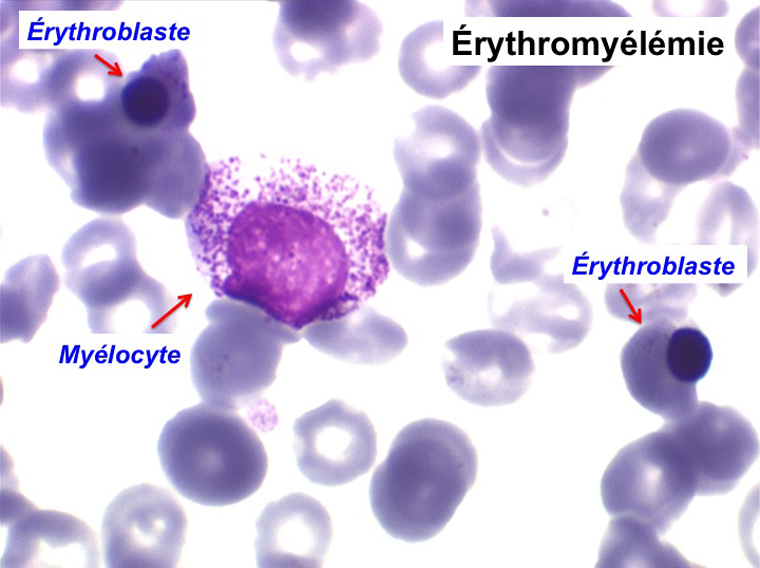
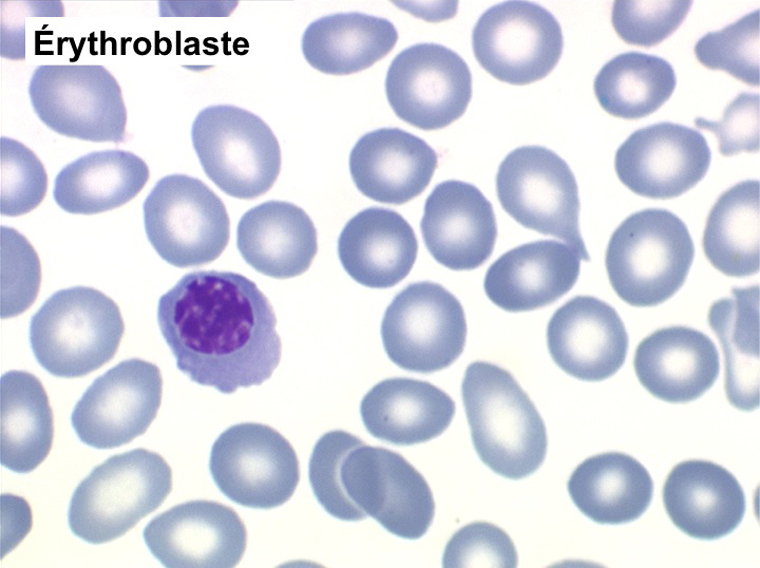
Une myélémie se définit par le passage dans le sang d'éléments immatures de la lignée granuleuse. Elle peut s'accompagner du passage d'érythroblastes, on parle alors d'érythromyélémie.
.jpg)
Les myélémies peuvent se diviser en trois groupes :
• les myélémies modérées et transitoires, généralement bénignes,
• les myélémies importantes et persistantes, généralement malignes,
• les érythromyélémies.
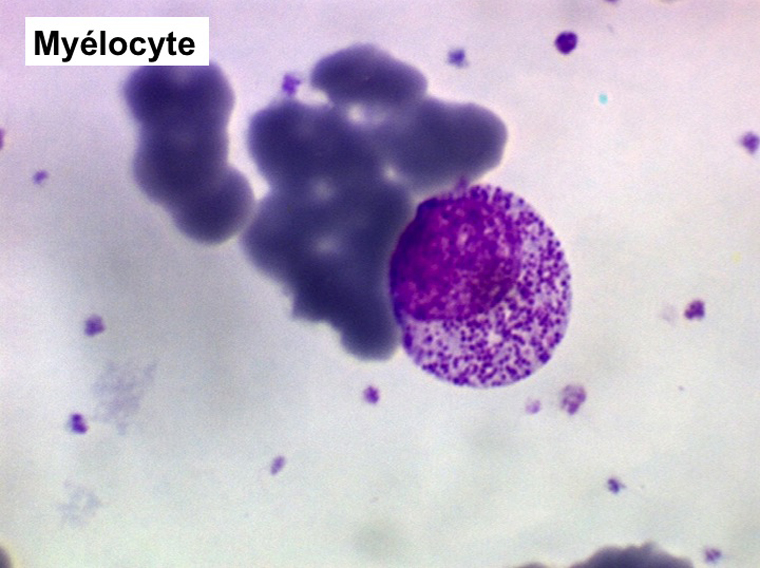
(1) – Les myélémies modérées et transitoires sont faites essentiellement de métamyélocytes et de quelques myélocytes qui habituellement ne dépassent pas au total 5% de l'ensemble des leucocytes circulants. Elles sont réactionnelles à une polynucléose infectieuse, une anémie régénérative (par hémorragie ou hémolyse), une sortie d'aplasie.
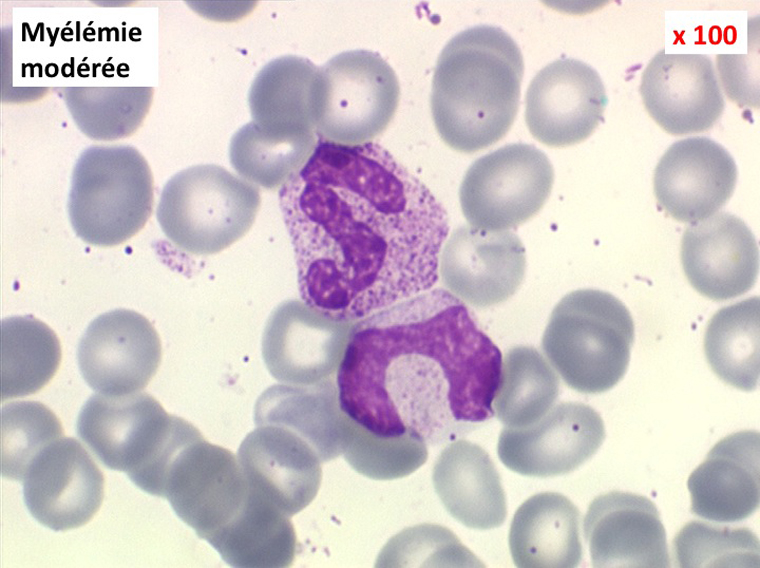
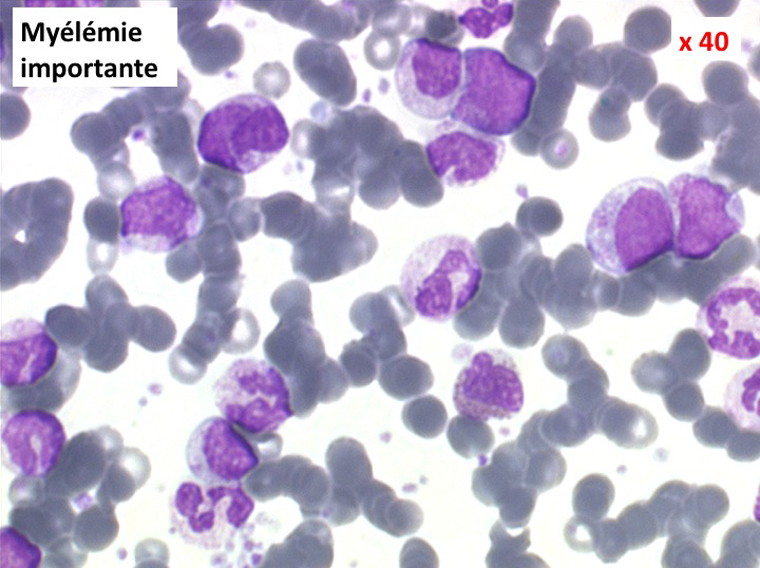
(2) – Les myélémies importantes et persistantes sont faites de tous les précurseurs granuleux, du myéloblaste au métamyélocyte, dont l'ensemble dépasse le plus souvent 10% des leucocytes. Elles peuvent s'accompagner de déformations des hématies. Elles traduisant une maladie maligne de la moelle osseuse :
• syndromes myéloprolifératifs (leucémie myéloïde chronique ou ostéo-myélosclérose),
• myélodysplasies,
• métastase médullaire.
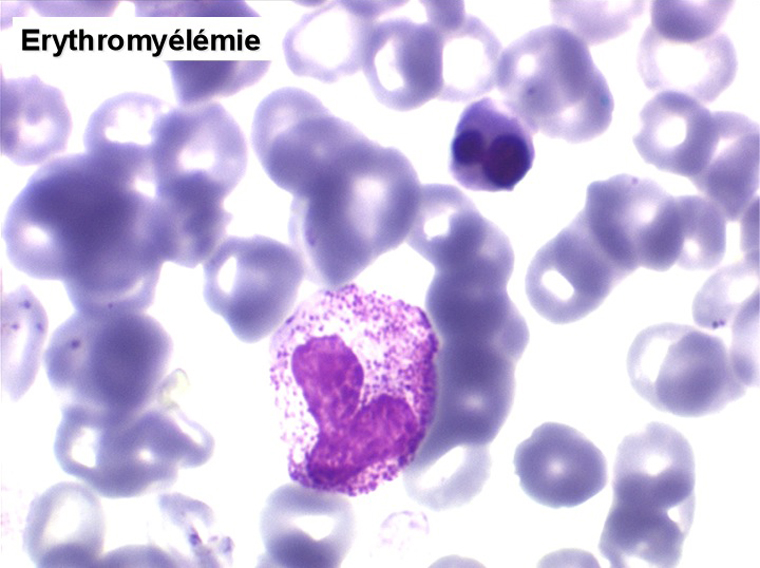
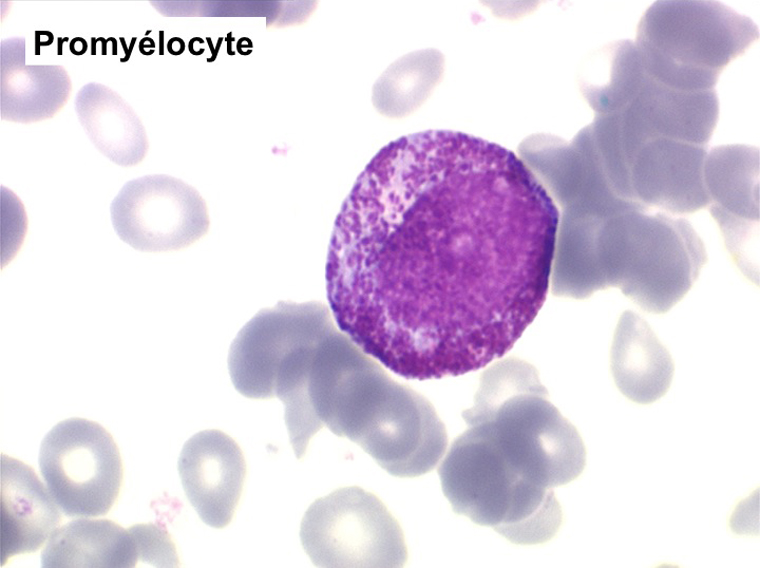
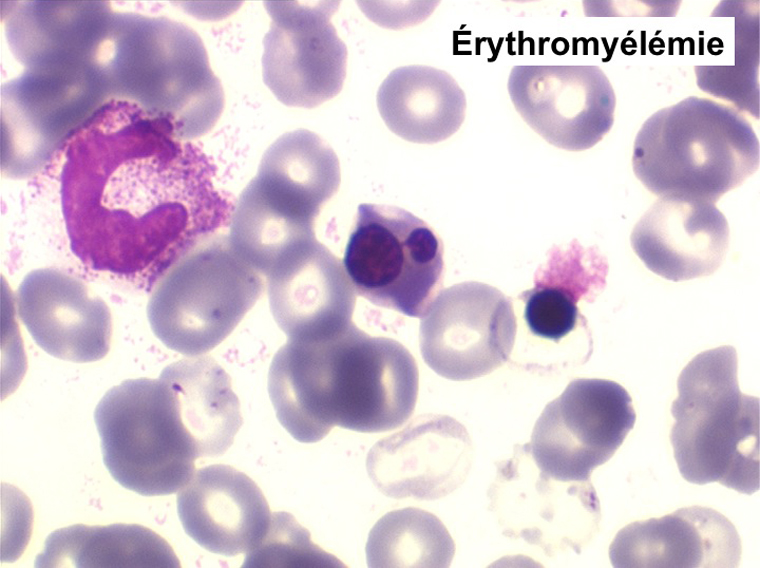
(3) – Les érythromyélémies sont définies comme le passage dans le sang de précurseurs des deux lignées, granuleuse (surtout des métamyélocytes et des myélocytes) et érythrocytaire (surtout érythroblastes acidophiles et polychromatophiles). Si le nombre d'érythroblastes circulants est important, ils doivent être défalqués du nombre total de leucocytes comptés par l'automate puisque celui-ci les comptabilise comme des globules blancs. L'origine d'une érythromyélémie dépend du caractère de l'anémie qui lui est associée :
• Si l'anémie est non régénérative il peut s'agir d'une ostéo-myélosclérose, d'une myélodysplasie (anémie réfractaire avec excès de blastes), ou d'une érythroleucémie aiguë (LAM6).
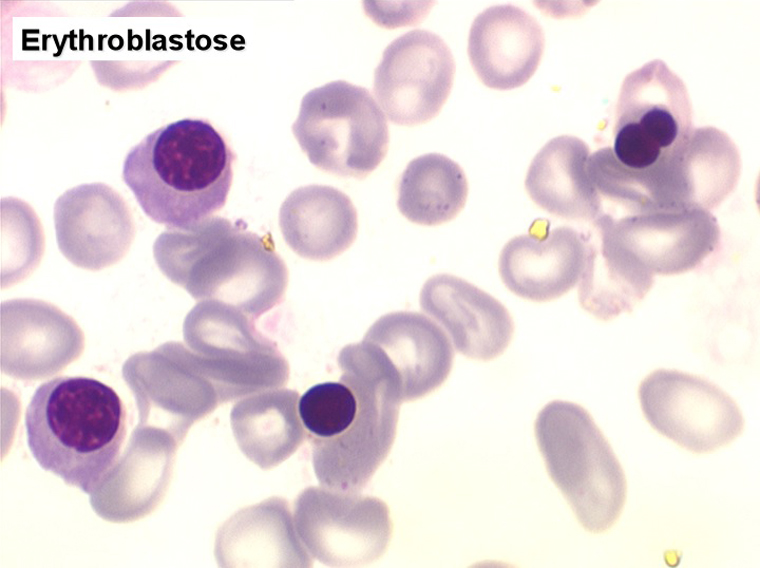
• Si l'anémie est fortement régénérative il s'agit d'une poussée d'anémie hémolytique dont l'illustration la plus probante est l'érythroblastose néonatale par incompatibilité fœto-maternelle.
• Si l'anémie est faiblement régénérative il faut penser à une métastase médullaire.
Les myélémies secondaires aux myélodysplasies seront décrites dans le chapitre correspondant (« Lignée granuleuse » → « Myélodysplasies »). Nous traiterons ici des myélémies des métastases médullaires et des syndromes myéloprolifératifs.
2 – Les métastases médullaires.
Les métastases médullaires s'observent préférentiellement dans certains cancers :
• cancers dits « ostéophiles » : rein, sein, prostate, thyroïde,
• cancer du poumon à petites cellules,
• mélanome malin,
• neuroblastome.
Outre l'érythromyélémie, les manifestations sanguines des métastases médullaires sont une pancytopénie par insuffisance médullaire mais remarquable par le fait que l'anémie, que l'on s'attendrait à voir non régénérative, est au contraire régénérative avec un taux de réticulocytes (modérément) augmenté. Ceci est dû à l'existence d'une hémolyse, les produits de dégradation provenant de la lyse de l'importante masse tumorale sont toxiques pour les globules rouges et diminuent leur durée de vie. Ceci explique aussi la fréquente constatation d'un taux de schizocytes élevé dans le sang.
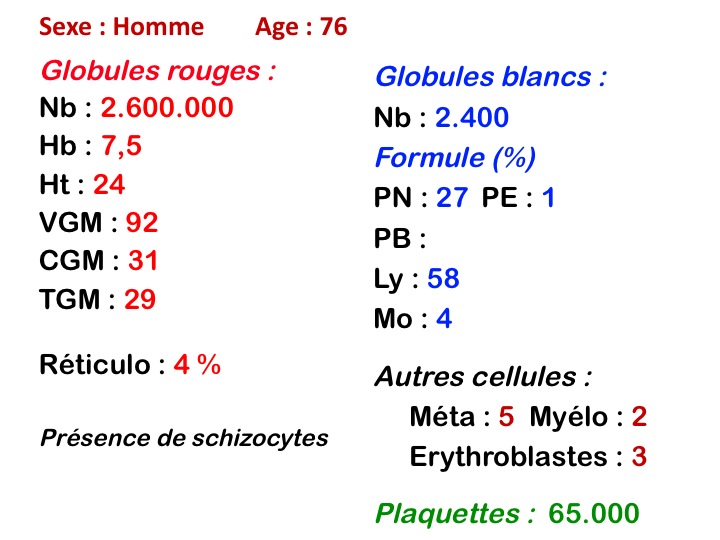
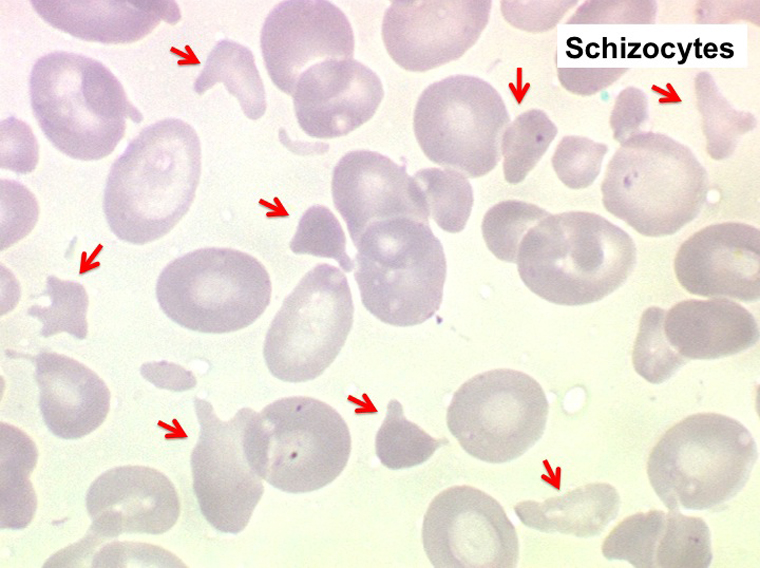
La preuve de la métastase est apportée par l'examen de la moelle osseuse prélevée par ponction, ou mieux par biopsie. La ponction médullaire montre des amas de cellules étrangères, formant souvent un syncytium de noyaux de tailles diverses (anisocytose nucléaire). Ces amas cellulaires doivent être recherchés à un faible grossissement (x 20). En dehors du mélanome malin, facilement identifiable par les dépôts de mélanine intracellulaires, il n'est pas possible de déterminer le siège de la tumeur sur le seul aspect morphologique des cellules métastatiques.
.jpg)
.jpg)
.jpg)
.jpg)
3 - Les syndromes myéloprolifératifs
Les syndromes myéloprolifératifs (SMP) sont des proliférations malignes chroniques d'une ou plusieurs lignées cellulaires de la moelle osseuse. Ces différents états pathologiques peuvent s'intriquer et s'enchainer. Ils ont tous la possibilité de se terminer en leucémie aiguë ou en sclérose médullaire, les uns fréquemment les autres exceptionnellement. On individualise dans les SMP quatre maladies différentes :
1. la leucémie myéloïde chronique (LMC)
2. la polyglobulie primitive,
3. l'ostéo-myélosclérose primitive (OMS),
4. la thrombocytémie primitive.
(1) – La leucémie myéloïde chronique (LMC)
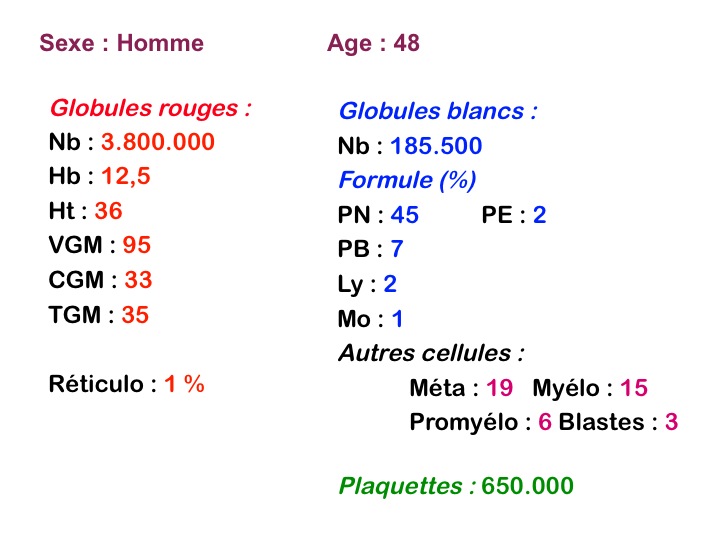
La maladie étant pendant longtemps asymptomatique, la NFS initiale est habituellement caractéristique montrant une hyperleucocytose très importante avec une myélémie non moins intense, si bien que l'aspect de la lame de sang est celui d'une moelle, les érythroblastes en moins. Une thrombocytose est fréquente, mais il n'y a pas d'anémie. Dans certaines formes on trouve dans le sang un excès d'éosinophiles ou plus souvent de basophiles, sans que cette particularité ait une incidence pronostique. Il est nécessaire d'apprécier et de suivre le taux sanguin de myéloblastes et de blastes indifférenciés compte tenu du risque de leucémie aiguë secondaire.
.jpg)
.jpg)
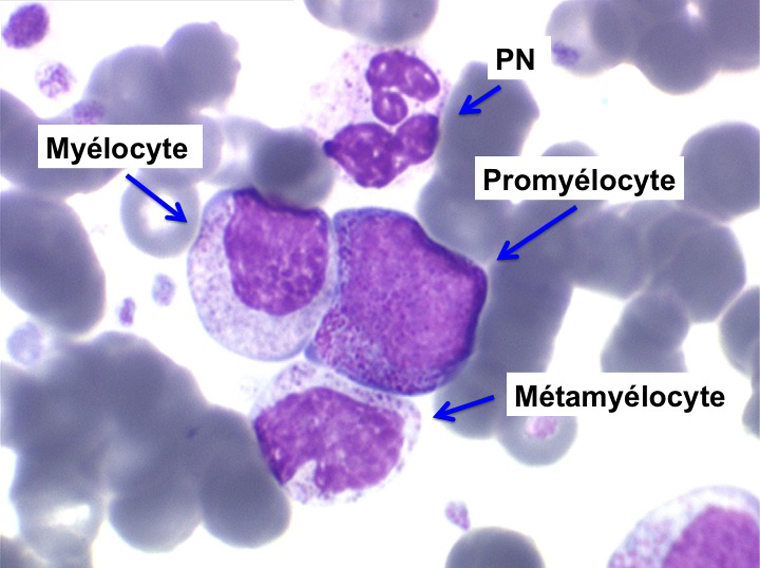
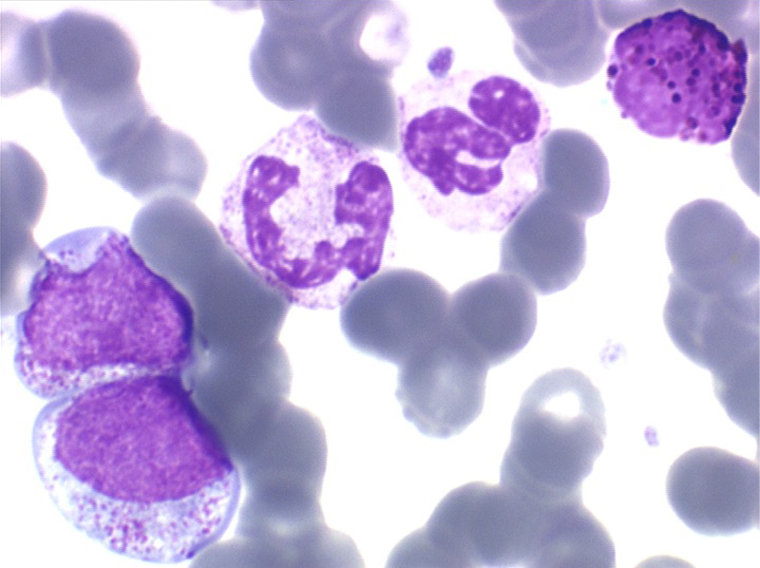
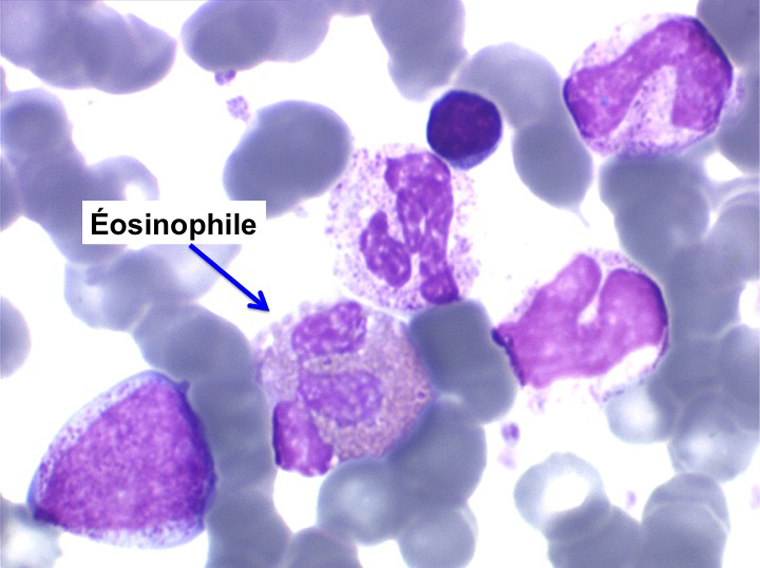

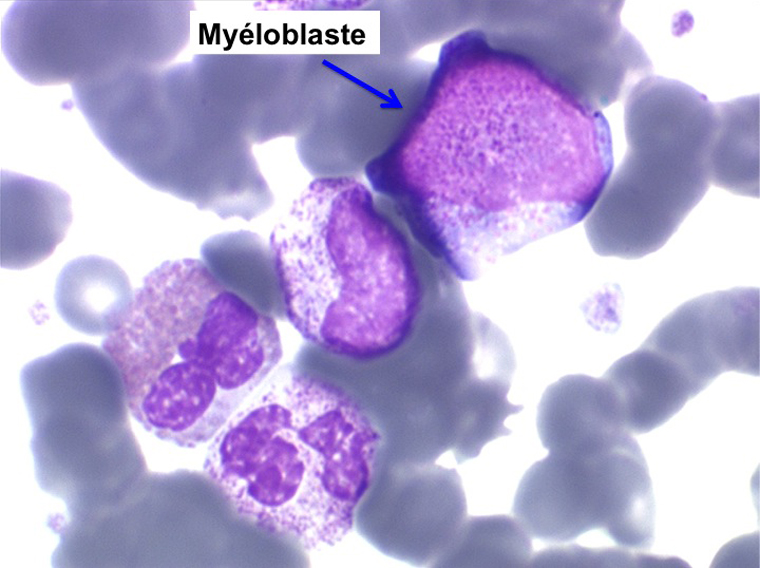
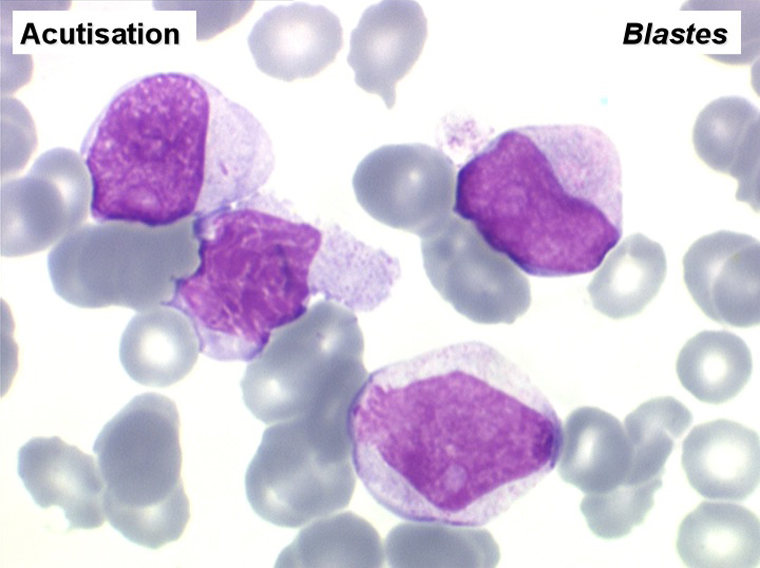
L'examen de la moelle osseuse n'a pas d'intérêt car il ne fait que montrer une « hyper-moelle » granuleuse. Une biopsie peut être utile pour rechercher une sclérose de mauvais pronostic. L'étude cytologique de la moelle est cependant essentielle pour établir un caryotype, si le sang ne le permet pas. La LMC est en effet caractérisée par une anomalie moléculaire spécifique, la translocation [ t(9q+;22q-) ] donnant un chromosome anormal dit de Philadelphie (Ph1) responsable de la maladie. À son stade chronique la maladie est bien supportée mais le risque est dans la survenue, le plus souvent brutale, d'une leucémie aiguë secondaire (l'acutisation), qui s'observe dans plus de 70% des cas si la maladie n'est pas traitée. Cette leucémie aiguë est dans 80% des cas myéloïde et dans 20% lymphoïde. Le traitement actuel, fondé sur le blocage de l'activité biologique du gène anormal, permet d'éviter cette redoutable complication.
(2) – La polyglobulie primitive (Maladie de Vaquez)
La polyglobulie est définie par une augmentation du taux d'hémoglobine, au dessus de 18 g/dl chez l'homme, de 16 g/dl chez la femme. Cette augmentation de l'hémoglobine va de pair avec une augmentation du nombre de globules rouges (polycytémie). L'examen de la lame de sang ne montre pas d'anomalie morphologique des globules rouges. Une légère hypochromie est possible mais s'observe surtout si le traitement comporte des saignées. L'augmentation des globules rouges peut s'associer à une polynucléose et à une thrombocytose, preuves de l'intense activité médullaire. De même on peut observer une myélémie, mais qui reste discrète, habituellement <5%. Ces stigmates de l'activité médullaire sont un bon signe de diagnostic différentiel avec les polyglobulies secondaires où l'augmentation des hématies reste isolée. Les formes de maladie de Vaquez où persistent polyglobulie importante, polynucléose, thrombocytose et myélémie doivent cependant être suspectées d'évolution rapide vers une myélosclérose.

Le plus important devant une polyglobulie primitive est de bien prendre en compte le taux de l'hématocrite. En effet celui-ci est le reflet direct de l'hyperviscosité sanguine qui augmente de façon exponentielle dans la polyglobulie. Au dessus de 60% d'hématocrite la viscosité sanguine atteint un degré tel (celle de la gelée de groseille !) que le malade court un risque vital. Il s'agit donc d'une urgence hématologique. Le malade doit être saigné au plus vite pour être mis à l'abri d'un accident de thrombose vasculaire, cérébrale ou cardiaque.

Pour différencier polyglobulie primitives et polyglobulies secondaires ou fausses polyglobulies par hémoconcentration, d'autres examens que la simple NFS peuvent s'avérer nécessaires tels que la masse sanguine, la saturation du sang en oxygène, la culture de moelle.
Les polyglobulies secondaires ont été traitées dans un autre chapitre (cf. « Sangs pathologiques » → « Globules rouges » → « Polyglobulies »).
L'évolution de la maladie de Vaquez est lente, la survie moyenne s'étale sur 10 à 15 ans. Une surveillance et un traitement corrects de la polyglobulie, en se basant sur l'hématocrite, permettent d'éviter les complications majeures de la maladie que sont les thromboses vasculaires. Cependant l'apparition progressive d'une myélosclérose, donnant un tableau d'OMS secondaire, est toujours possible. De même le risque leucémique n'est jamais exclu, même s'il n'excède pas 2% des polyglobulies primitives.
(3) – L'ostéo-myélosclérose primitive (OMS)
Comme son nom l'indique l'ostéo-myélosclérose primitive est une sclérose de la moelle osseuse avec une ossification progressive qui étouffe les logettes médullo-sanguines et aboutit à une disparition du tissu hématopoïétique. L'hématopoïèse ne pouvant plus se faire dans la moelle, le foie et la rate prennent le relai (comme chez le fœtus) et deviennent énormes. C'est cette hématopoïèse extra-médullaire qui a donné son autre nom à la maladie, souvent appelée en France « splénomégalie myéloïde ».
La NFS montre une hyperleucocytose de l'ordre de 10.000/µl à 20.000/µl, n'excédant pas 50.000/µl. Elle s'accompagne d'une anémie non régénérative et d'une thrombopénie. La myélémie est nette mais différente de celle de la LMC avec un pourcentage important de blastes sans que cela ait une incidence pronostique et la présence d'érythroblastes (érythromyélémie).


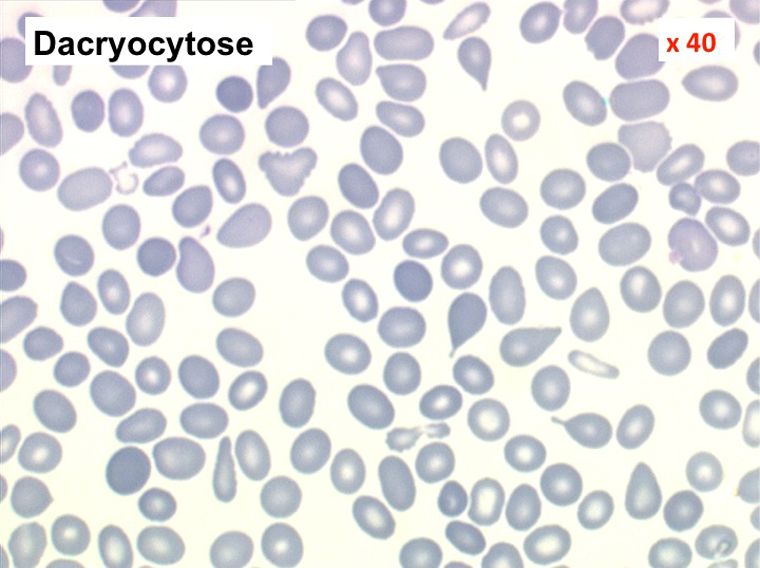
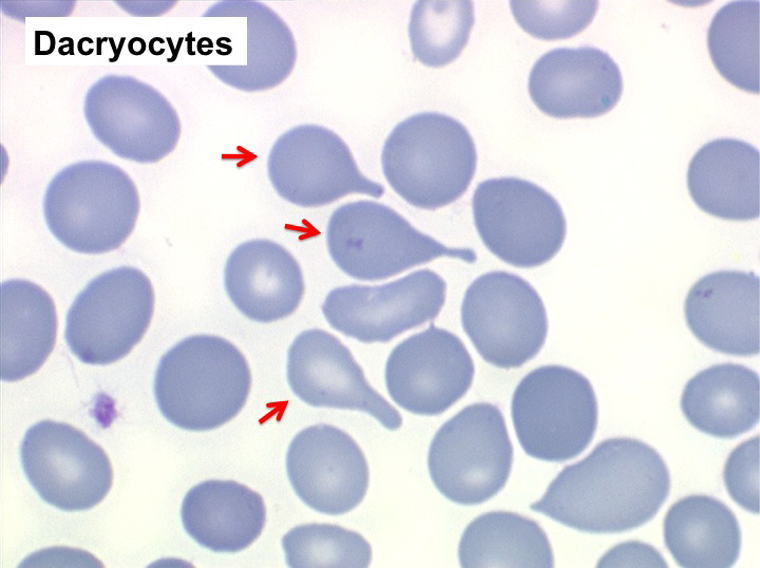
Mais l'anomalie cytologique dominante qui doit faire évoquer le diagnostic est la déformation des globules rouges. La plus caractéristique est la dacryocytose (hématies en poires, en larmes, en gouttes), mais elle peut s'associer à des anomalies multiples (ovalocytes, schizocytes, anisocytose). L'association d'une myélémie et de déformations des hématies doit immédiatement évoquer le diagnostic d'OMS.

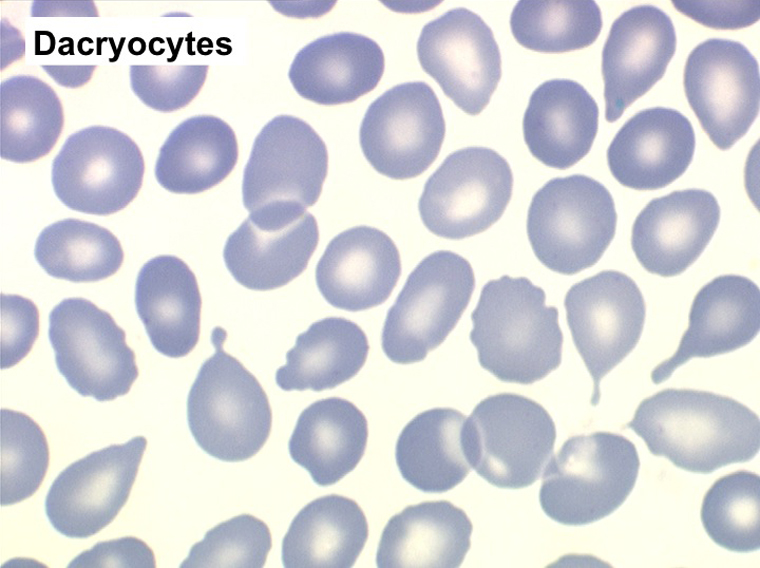
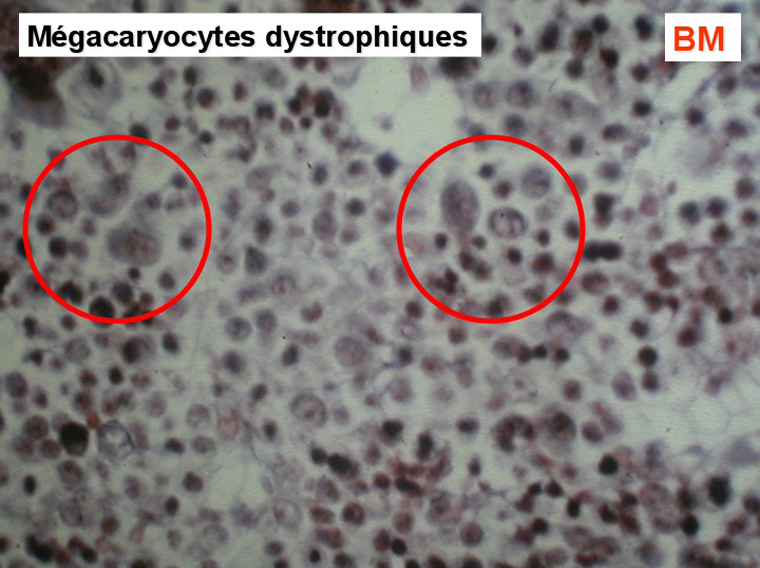
L'examen de la moelle osseuse par ponction est habituellement impossible tant l'os est dur. Seule la biopsie apporte la preuve de la fibrose et de l'ostéosclérose, les rares cellules hématopoïétiques observées sont isolées les unes des autres par la fibrose. Les mégacaryocytes sont très nombreux et dystrophiques, ils jouent un grand rôle dans le développement de la fibrose.
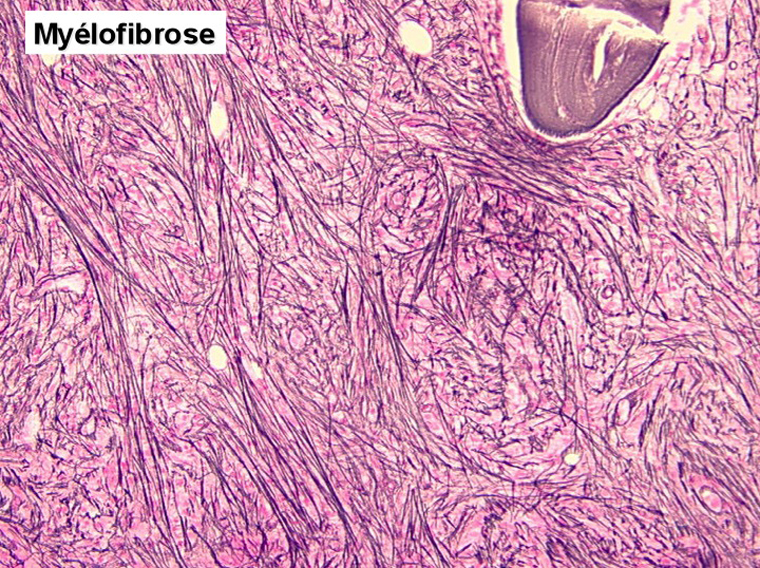

La métaplasie myéloïde de la rate et du foie pourrait être mise en évidence par la ponction de ces organes dont le frottis ressemble à de la moelle osseuse avec toutes les lignées présentes. Mais ces ponctions ne sont pas sans risque et on a actuellement d'autres moyens (notamment isotopiques) de mettre en évidence cette métaplasie. L'évolution est fatale en 3 à 5 ans par cachexie, aplasie ou beaucoup plus rarement leucémie aiguë secondaire.
(4) – La thrombocytémie primitive
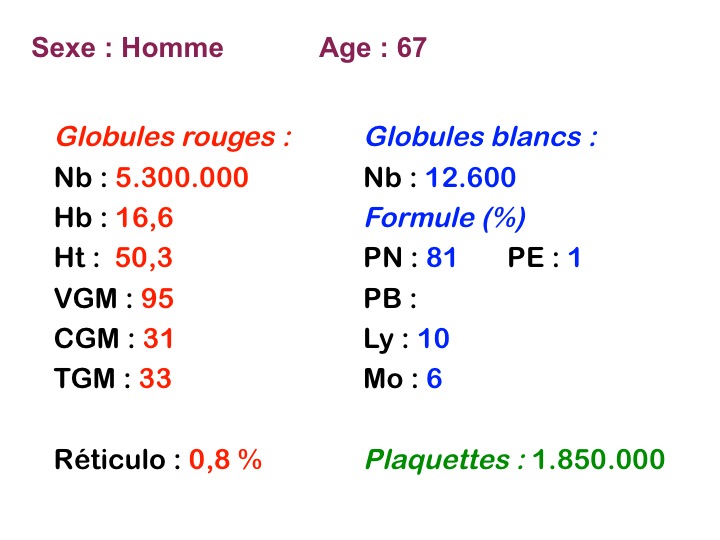
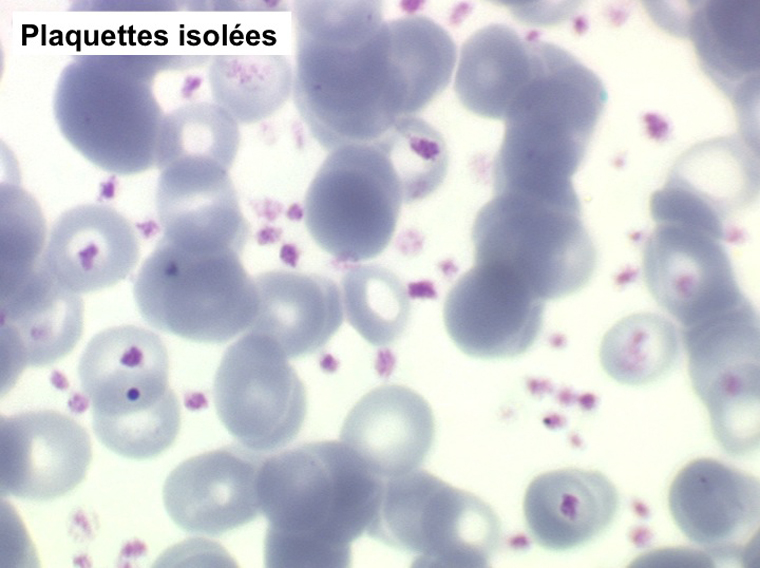
La thrombocytémie primitive (aussi appelée thrombocytémie essentielle) est caractérisée par une importante augmentation du taux de plaquettes sanguines, supérieur à 600.000/µl, le plus souvent supérieur à 1 million par microlitre. La morphologie des plaquettes est normale mais la thrombocytémie peut avoir deux aspects différents : plaquettes isolées, bien séparées les unes des autres, ou plaquettes en amas. Dans ce dernier cas l'automate peut sous-estimer le nombre réel de plaquettes en comptabilisant les amas comme des leucocytes. Il convient donc de respecter la règle qui veut que l'on vérifie toujours sur lame la quantité et la répartition des plaquettes lorsque le taux de celles-ci pose problème.
La thrombocytose est le plus souvent isolée, mais la NFS peut montrer une légère polynucléose et un taux d'hémoglobine dans les limites supérieures de la normale. En revanche une myélémie est rare, éventuellement réduite à quelques métamyélocytes.
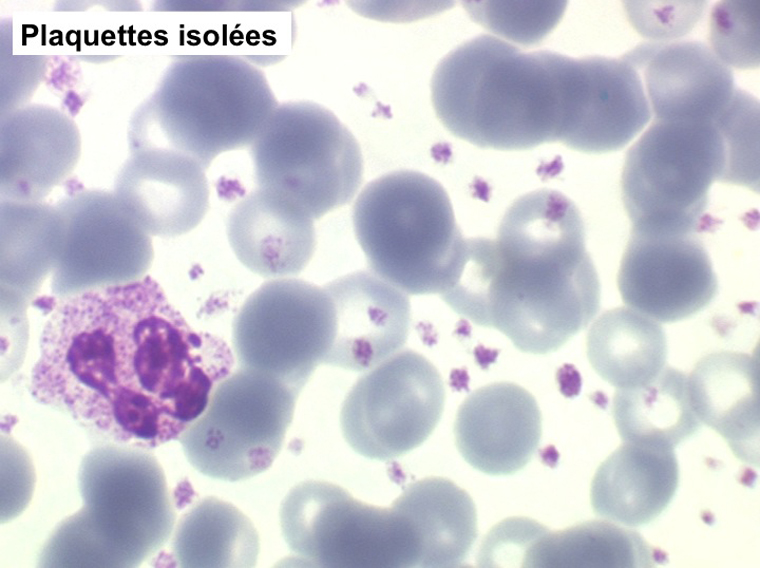
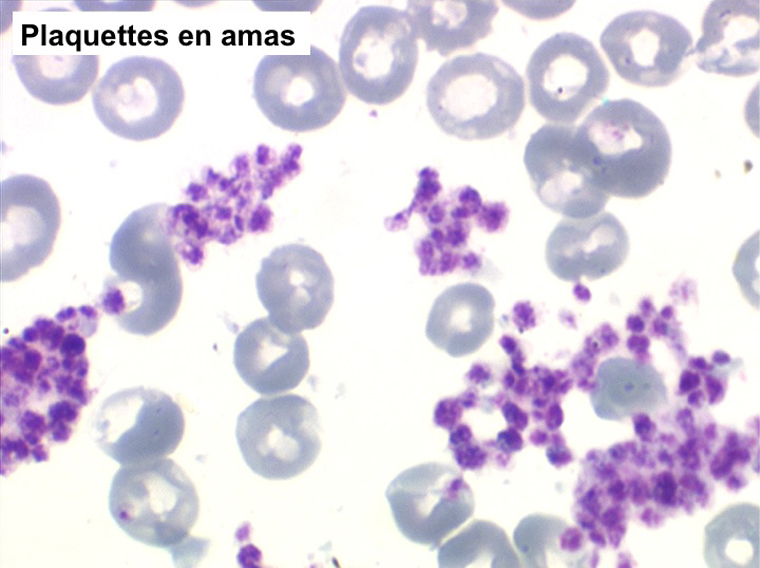
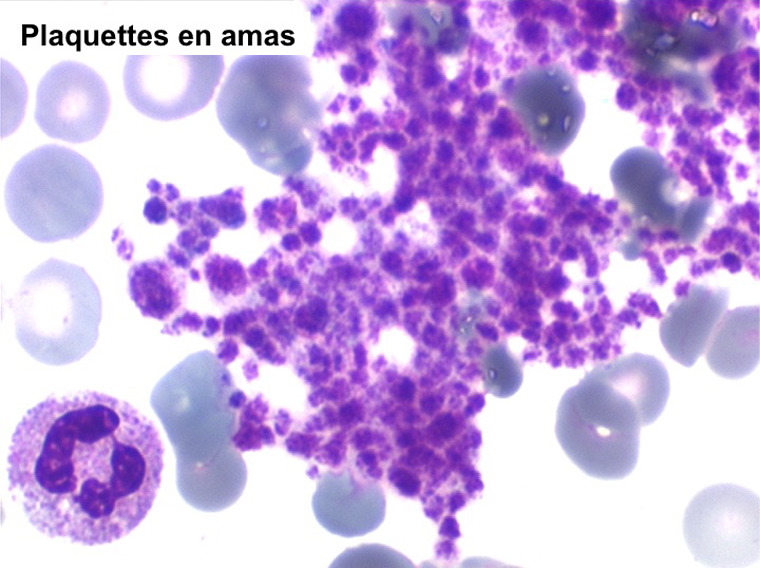
Le myélogramme n'a guère d'intérêt pour confirmer le diagnostic. Il montrerait la grande richesse de la moelle en mégacaryocytes, souvent groupés en amas, dont la morphologie est normale. La biopsie médullaire doit être évitée car il existe un risque hémorragique, la thrombocytose étant responsable d'un déficit de l'hémostase primaire (temps de saignement allongé).
.jpg)
Une forme particulière de thrombocytémie est le syndrome 5q qui s'apparente à l'anémie réfractaire (cf. le chapitre « Lignée granuleuse » → « Myélodysplasies »). La thrombocytose y est importante (> 1 million/µl) avec dans la moelle des mégacaryocytes anormaux, non polylobés. Il s'y associe une anémie le plus souvent macrocytaire.
L'augmentation persistante des plaquettes au dessus du million est le meilleur critère pour différencier la thrombocytémie primitive des thrombocytoses secondaires, mais ce diagnostic différentiel n'est pas toujours facile quand la thrombocytose est modérée. On peut alors s'aider de la cytogénétique et rechercher la mutation du gène JAK2, qui n'est cependant retrouvée que dans 70% des thrombocytémies primitives.
L'évolution de la thrombocytémie primitive est très longue avec possibilité de complications hémorragiques ou thrombotiques. La transformation en leucémie aiguë est très rare (<5%) de même qu'une myélosclérose progressive
Dans les thrombocytoses secondaires le taux de plaquettes se situe entre 500.000/µl et 800.000/µl, rarement plus ou alors transitoirement. Les principales causes de thrombocytoses secondaires sont :
• soit réactionnelles à une splénectomie (il y a alors sur lame de nombreux corps de Jolly) ou à une anémie régénérative,
• soit malignes au cours de cancers (syndrome paranéoplasique).